- 2-Hydroxyglutaric aciduria
-
2-Hydroxyglutaric aciduria Classification and external resources 
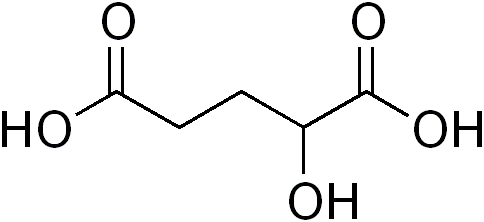
Alpha-Hydroxyglutaric acidOMIM 600721 236792 DiseasesDB 34515 34514 2-hydroxyglutaric aciduria is a rare, autosomal recessive neurometabolic disorder characterized by the significant elevation of urinary levels of hydroxyglutaric acid.
Contents
Classification
 Both forms of 2-Hydroxyglutaric aciduria have an autosomal recessive pattern of inheritance.
Both forms of 2-Hydroxyglutaric aciduria have an autosomal recessive pattern of inheritance.
2-hydroxyglutaric aciduria is an organic aciduria, and has two distinct stereoisomeric variants:
L-2-hydroxyglutaric aciduria
The L-2 form is more common, severe, and mainly affects the central nervous system. The basal ganglia are affected, and cystic cavitations in the white matter of the brain are common, beginning in infancy. This form is chronic, with early symptoms such as hypotonia, tremors, and epilepsy declining into spongiform leukoencephalopathy, muscular choreodystonia, mental retardation, and psychomotor regression.[1]
It is associated with L2HGDH.[2]
D-2-hydroxyglutaric aciduria
The D2 form is rare, with symptoms including macrocephaly, cardiomyopathy, mental retardation, hypotonia, and cortical blindness.[3]
It can be associated with D2HGDH.
See also
References
- ^ Seijo-Martinez M, Navarro C, Castro del Rio M, Vila O, Puig M, Ribes A, Butron M (2005). "L-2-hydroxyglutaric aciduria: clinical, neuroimaging, and neuropathological findings". Arch Neurol. 62 (4): 666–670. doi:10.1001/archneur.62.4.666. PMID 15824270.
- ^ Topçu M, Jobard F, Halliez S, et al. (November 2004). "L-2-Hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1". Hum. Mol. Genet. 13 (22): 2803–11. doi:10.1093/hmg/ddh300. PMID 15385440. http://hmg.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=15385440.
- ^ Nyhan WL, Shelton GD, Jakobs C, Holmes B, Bowe C, Curry CJ, Vance C, Duran M, Sweetman L (1995). "D-2-hydroxyglutaric aciduria". J Child Neurol. 10 (2): 137–142. doi:10.1177/088307389501000216. PMID 7782605.
Inborn error of amino acid metabolism (E70–E72, 270) K→acetyl-CoA Lysine/straight chainHypertryptophanemiaG G→pyruvate→citrateG→glutamate→
α-ketoglutarateGlutamate/glutamineG→fumarateType II tyrosinemia · Type III tyrosinemia/Hawkinsinuria · Alkaptonuria/Ochronosis · Type I tyrosinemiaTransport/
IE of RTTOther Categories:- Inborn errors of metabolism
- Autosomal recessive disorders
- Rare diseases

Wikimedia Foundation. 2010.