- Creutzfeldt–Jakob disease
-
Creutzfeldt–Jakob disease Classification and external resources 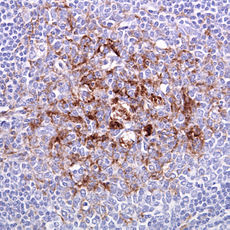
Tonsil biopsy in variant CJD. Prion Protein immunostaining.ICD-10 A81.0, F02.1 ICD-9 046.1 OMIM 123400 DiseasesDB 3166 eMedicine neuro/725 MeSH D007562 Creutzfeldt–Jakob disease or CJD (pronounced /ˈkrɔɪtsfɛlt ˈjɑːkoʊb/,[1] "KROITS-felt YA-kob") is a degenerative neurological disorder (brain disease) that is incurable and invariably fatal.[2] CJD is at times called a human form of mad cow disease, given that bovine spongiform encephalopathy is believed to be the cause of variant Creutzfeldt–Jakob disease in humans.[3]
CJD is the most common among the types of transmissible spongiform encephalopathy found in humans.[4] In CJD, the brain tissue develops holes and takes on a sponge-like texture. This is due to a type of infectious protein called a prion. Prions are misfolded proteins which replicate by converting their properly folded counterparts.
Contents
Classification
Types of CJD include:
- sporadic (sCJD)[5]
- variant (vCJD)[6] This type is more likely to be acquired. It can be iatrogenic.[7] It was first identified in 1996.[8]
- familial (fCJD)[9]
Signs and symptoms
The first symptom of CJD is rapidly progressive dementia, leading to memory loss, personality changes and hallucinations. This is accompanied by physical problems such as speech impairment, jerky movements (myoclonus), balance and coordination dysfunction (ataxia), changes in gait, rigid posture, and seizures. The duration of the disease varies greatly, but sporadic (non-inherited) CJD can be fatal within months or even weeks (Johnson, 1998). In some people, the symptoms can continue for years. In most patients, these symptoms are followed by involuntary movements and the appearance of an atypical diagnostic electroencephalogram tracing. Most victims die 6 months after initial symptoms appear, often of pneumonia due to impaired coughing reflexes. About 15% of patients survive 2 or more years.[10]
The symptoms of CJD are caused by the progressive death of the brain's nerve cells, which is associated with the build-up of abnormal prion proteins forming amyloids. When brain tissue from a CJD patient is examined under a microscope, many tiny holes can be seen where whole areas of nerve cells have died. The word "spongiform" in "transmissible spongiform encephalopathies" refers to the sponge-like appearance of the brain tissue.
Cause
Transmissible spongiform encephalopathy diseases are caused by prions. The diseases are thus sometimes called prion diseases. Other prion diseases include Gerstmann–Sträussler–Scheinker syndrome (GSS), fatal familial insomnia (FFI) and kuru in humans, as well as bovine spongiform encephalopathy (BSE, commonly known as mad cow disease) in cattle, chronic wasting disease (CWD) in elk and deer, and scrapie in sheep. Alpers' syndrome in infants is also thought to be a transmissible spongiform encephalopathy caused by a prion.[11][12]
The prion that is believed to cause Creutzfeldt–Jakob exhibits at least two stable conformations. One, the native state, is water-soluble and present in healthy cells. As of 2007[update], its biological function is presumably in transmembrane transport or signaling. The other conformational state is relatively water-insoluble and readily forms protein aggregates.
People can also acquire CJD genetically through a mutation of the gene that codes for the prion protein (PRNP). This occurs in only 5–10% of all CJD cases.
The CJD prion is dangerous because it promotes refolding of native proteins into the diseased state.[citation needed] The number of misfolded protein molecules will increase exponentially and the process leads to a large quantity of insoluble protein in affected cells. This mass of misfolded proteins disrupts cell function and causes cell death. Mutations in the gene for the prion protein can cause a misfolding of the dominantly alpha helical regions into beta pleated sheets. This change in conformation disables the ability of the protein to undergo digestion. Once the prion is transmitted, the defective proteins invade the brain and are produced in a self-sustaining feedback loop.
Stanley B. Prusiner of the University of California, San Francisco (UCSF) was awarded the Nobel Prize in physiology or medicine in 1997 for his discovery of prions. For more than a decade, Yale University neuropathologist Laura Manuelidis has been challenging this explanation for the disease. In January 2007, she and her colleagues published an article in the Proceedings of the National Academy of Science and reported that they have found a virus-like particle (but without finding nucleic acids so far) in less than 10% of the cells a scrapie-infected cell line and in a mouse cell line infected by a human CJD agent.[13]
Transmission
The defective protein can be transmitted by contaminated harvested human brain products, Immunoglobulins (IVIG), corneal grafts, dural grafts or electrode implants (acquired or iatrogenic form: iCJD); it can be inherited (hereditary or familial form: fCJD); or it may appear for the first time in the patient (sporadic form: sCJD). In the hereditary form, a mutation occurs in the gene for PrP, PRNP. Ten to 15 percent of CJD cases are inherited. (CDC)
The disease has also been shown to result from use of Human Growth Hormone obtained from the pituitary glands of persons who died from Creutzfeldt–Jakob Disease,[14] though the known incidence of this cause is (as of April 2004) quite small. The risk of infection via cadaveric HGH in the US ceased when the medication was withdrawn in 1985.
It is thought[citation needed] that humans can contract the disease by consuming material from animals infected with the bovine form of the disease. The only suspected cases to arise thus far have been vCJD, although there are fears—based on animal studies—that consuming beef or beef products containing prion particles can also cause the development of classic CJD. When BSE material infects humans, the resulting disease is known as (new) variant CJD (nvCJD).[12]
Cannibalism has also been implicated as a transmission mechanism for abnormal prions, causing the disease known as kuru, once found primarily among women and children of the Fore people in Papua New Guinea. While the men of the tribe ate the body of the deceased and rarely contracted the disease, the women and children, who ate the less desirable body parts, including the brain, were 8 times more likely than men to contract kuru from infected tissue.
Prions, the infectious agent of CJD, may not be inactivated by means of routine surgical instrument sterilization procedures. The World Health Organization and the US Centers for Disease Control and Prevention recommend that instrumentation used in such cases be immediately destroyed after use; short of destruction, it is recommended that heat and chemical decontamination be used in combination to process instruments that come in contact with high-infectivity tissues. No cases of iatrogenic transmission of CJD have been reported subsequent to the adoption of current sterilization procedures, or since 1976.[15][16][17] Copper–hydrogen peroxide has been suggested as an alternative to the current recommendation of sodium hydroxide or sodium hypochlorite.[18] Thermal depolymerization also destroys prions in infected organic and inorganic matter, since the process chemically attacks protein at the molecular level.
Blood donor restrictions
In 2004 a new report published in the Lancet medical journal showed that vCJD can be transmitted by blood transfusions.[19] The finding alarmed healthcare officials because a large epidemic of the disease could result in the near future. There is no test to determine if a blood donor is infected with vCJD. In reaction to this report, the UK government banned anyone who had received a blood transfusion since January 1980 from donating blood.[20] From 1999 there has been a ban in the UK for using UK blood to manufacture fractional products such as albumin.[21]
On May 28, 2002, the United States Food and Drug Administration instituted a policy that excludes from donation anyone who spent at least six months in certain European countries, (or three months in the United Kingdom), from 1980 to 1996. Given the large number of U.S. military personnel and their dependents residing in Europe, it was expected that over 7% of donors would be deferred due to the policy. Later changes to this policy have relaxed the restriction to a cumulative total of five years or more of civilian travel in European countries (six months or more if military). The three-month restriction on travel to the UK, however, has not been changed.[22]
The American Red Cross' policy is as follows: During the period January 1, 1980, to December 31, 1996, spending a total time of three months or more in the Channel Islands, England, the Falkland Islands, the Isle of Man, Gibraltar, Northern Ireland, Scotland, and Wales precludes individuals from donating. Moreover, spending a total time of five years or more after January 1, 1980 (to present), in the above-mentioned countries and/or any country in Europe (except the former USSR), also precludes donation. People with a biologic relative who has been diagnosed with CJD or vCJD are unable to donate. Biologic relative in this setting means mother, father, sibling, grandparent, aunt, uncle or children. (For complete listing, please go to Redcross.org)
A similar policy applies to potential donors to the Australian Red Cross' Blood Service, precluding people who have spent a cumulative time of six months or more in the United Kingdom between 1980 and 1996.
The Singapore Red Cross precludes potential donors who have spent a cumulative time of three months or more in the United Kingdom between 1980 and 1996.
In New Zealand, anyone who has lived in the UK, France or the Republic of Ireland for a total of six months or more between 1980 and 1996 is prohibited from donating blood.
Similar regulations are in place in Germany, where anyone who has spent six months or more living in the UK between January 1980 and December 1996 is permanently barred from donating blood.[23]
As of 1999, Health Canada announced a policy to defer individuals from donating blood if they have lived within the United Kingdom for one month or more from January 1, 1980, to December 31, 1996. In 2000, the same policy was applied to people who have resided in France, for at least three months from January 1980 to December 1996. Canada will not accept blood from a person who has spent more than six months in a Western European country since January 1, 1980.[24]
The Association of Blood Donors of Denmark precludes potential donors who have spent a cumulative time of at least 12 months in the United Kingdom between 1 January 1980 and 31 December 1996.
The Swiss Blutspendedienst SRK precludes potential donors who have spent a cumulative time of at least six months in the United Kingdom between 1 January 1980 and 31 December 1996.
In Poland, anyone who cumulatively spent six months or longer between 1 January 1980 and 31 December 1996 in the UK, Ireland or France is permanently barred from donating.[25]
In the Czech Republic, anyone who spent more than six months in the UK or France between the years 1980 and 1996 or received transfusion in the UK after the year 1980 is not allowed to donate blood.[26]
Sperm donor restrictions
In the U.S., the FDA has banned import of any donor sperm, motivated by a risk of Creutzfeldt–Jakob disease, inhibiting the once popular[27] import of, for example, Scandinavian sperm. The risk, however, is not known, since artificial insemination has not been studied as a route of transmission.[28][28]
Diagnosis
The diagnosis of CJD is suspected when there are typical clinical symptoms and signs such as rapidly progressing dementia with myoclonus. Further investigation can then be performed to support the diagnosis including
- Electroencephalography — often has characteristic triphasic spikes
- Cerebrospinal fluid analysis for 14-3-3 protein
- MRI of the brain — often shows high signal intensity in the caudate nucleus and putamen bilaterally on T2-weighted images.
- Research in 2010 and 2011 identified a possible blood test for CJD. The test attempts to identify the prion responsible for the disease. However, it was unable to detect the prions in those in early stages of the disease.[29]
Diffusion Weighted Imaging (DWI) images are the most sensitive. In about 24% of cases DWI shows only cortical hyperintensity; in 68%, cortical and subcortical abnormalities; and in 5%, only subcortical anomalies.[30] The involvement of the thalamus can be found in sCJD, is even stronger and constant in vCJD.[31]
Clinical testing for CJD has always been an issue. Diagnosis has mostly been based on clinical and physical examination of symptoms. In recent years, studies have shown that the tumour marker Neuron-specific enolase (NSE) is often elevated in CJD cases, however its diagnostic utility is primarily seen when combined with a test for the 14-3-3 protein.[32] As of 2010[update], screening tests to identify infected asymptomatic individuals, such as blood donors, are not yet available, though methods have been proposed and evaluated.[33]
In 2010, A team from New York described detection of PrPSc even when initially present at only one part in one hundred billion (10−11) in brain tissue. The method combines amplification with a novel technology called Surround Optical Fiber Immunoassay (SOFIA) and some specific antibodies against PrPSc. After amplifying and then concentrating any PrPSc, the samples are labelled with a fluorescent dye using an antibody for specificity and then finally loaded into a micro-capillary tube. This tube is placed in a specially constructed apparatus so that it is totally surrounded by optical fibres to capture all light emitted once the dye is excited using a laser. The technique allowed detection of PrPSc after many fewer cycles of conversion than others have achieved, substantially reducing the possibility of artefacts, as well as speeding up the assay. The researchers also tested their method on blood samples from apparently healthy sheep that went on to develop scrapie. The animals’ brains were analysed once any symptoms became apparent. The researchers could therefore compare results from brain tissue and blood taken once the animals exhibited symptoms of the diseases, with blood obtained earlier in the animals’ lives, and from uninfected animals. The results showed very clearly that PrPSc could be detected in the blood of animals long before the symptoms appeared. After further development and testing, this method could be of great value in surveillance as a blood or urine-based screening test for CJD.[34][35]
In one third of patients with sporadic CJD, deposits of "prion protein (scrapie)," PrPSc, can be found in the skeletal muscle and/or the spleen.[citation needed] Diagnosis of vCJD can be supported by biopsy of the tonsils, which harbour significant amounts of PrPSc; however, biopsy of brain tissue is the definitive diagnostic test. Due to its invasiveness, biopsy will not be done if clinical suspicion is sufficiently high or low. A negative biopsy does not rule out CJD, since it may predominate in a specific part of the brain[36]
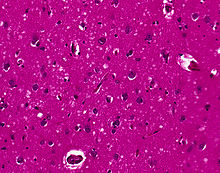 Spongiform change in CJD
Spongiform change in CJD
The classic histologic appearance is spongiform change in the gray matter: the presence of many round vacuoles from one to 50 micrometres in the neuropil, in all six cortical layers in the cerebral cortex or with diffuse involvement of the cerebellar molecular layer. These vacuoles appear glassy or eosinophilic and may coalesce. Neuronal loss and gliosis are also seen.[37] Plaques of amyloid-like material can be seen in the neocortex in new-variant CJD.
Unfortunately, vacuolization can be seen in other disease states. Diffuse cortical vacuolization occurs in Alzheimer's, and superficial cortical vacuolization occurs in ischemia and frontotemporal dementia. These vacuoles appear clear and punched-out. Larger vacuoles encircling neurons, vessels, and glia are a possible processing artifact.[36]
- Clinical and Pathologic Characteristics:[38]
Characteristic Classic CJD Variant CJD Median age at death 68 years 28 years Median duration of illness 4–5 months 13–14 months Clinical signs and symptoms Dementia; early neurologic signs Prominent psychiatric/behavioral symptoms; painful dysesthesias; delayed neurologic signs
Periodic sharp waves on electroencephalogram Often present Often absent Signal hyperintensity in the caudate nucleus and putamen on diffusion-weighted and FLAIR MRI Often present Often absent Pulvinar sign-bilateral high signal intensities on axial Fluid attenuated inversion recovery (FLAIR) MRI. Also posterior thalami involvement on sagittal T2 sequences Not reported Present in >75% of cases Immunohistochemical analysis of brain tissue Variable accumulation. Marked accumulation of protease-resistant prion protein Presence of agent in lymphoid tissue Not readily detected Readily detected Increased glycoform ratio on immunoblot analysis of protease-resistant prion protein
Not reported Marked accumulation of protease-resistant prion protein Presence of amyloid plaques in brain tissue May be present May be present - An abnormal signal in the posterior thalamus on T2- and diffusion-weighted images and fluid-attenuated inversion recovery sequences on brain magnetic resonance imaging (MRI); in the appropriate clinical context, this signal is highly specific for vCJD. (Source: CDC)
Treatment
As of 2011[update] no generally accepted treatment for CJD exists; the disease is invariably fatal and research continues. An experimental treatment was given to a Northern Irish teenager, Jonathan Simms, beginning in January 2003.[39] The medication, called pentosan polysulphate (PPS) and used to treat interstitial cystitis, is infused into the patient's lateral ventricle within the brain. PPS does not seem to stop the disease from progressing, and both brain function and tissue continue to be lost. However, the treatment is alleged to slow the progression of the otherwise untreatable disease, and may have contributed to the longer than expected survival of the seven patients who were studied.[40] The CJD Therapy Advisory Group to the UK Health Departments advises that data are not sufficient to support claims that pentosan polysulphate is an effective treatment and suggests that further research in animal models is appropriate.[41] A 2007 review of the treatment of 26 patients with PPS finds no proof of efficacy because of the lack of accepted objective criteria.[42]
Scientists have investigated using RNA interference to slow the progression of scrapie in mice. The RNA blocks production of the protein that the CJD process transforms into prions. This research is unlikely to lead to a human therapy for many years.[43]
Both amphotericin B and doxorubicin have been investigated as potentially effective against CJD, but as yet there is no strong evidence that either drug is effective in stopping the disease. Further study has been taken with other medical drugs, but none are effective. However, drugs to reduce suffering do exist, and include Valproate, an anticonvulsant, and Clonazepam, to reduce muscle jerks.[10]
Dr. Michael Geschwind, Dr. Bruce Miller and Dr. Stanley Prusiner from University of California, San Francisco are currently running a treatment trial for sporadic CJD using quinacrine, a medicine originally created for malaria. Pilot studies showed quinacrine permanently cleared abnormal prion proteins from cell cultures, but results have not yet been published on the clinical study.
Epidemiology
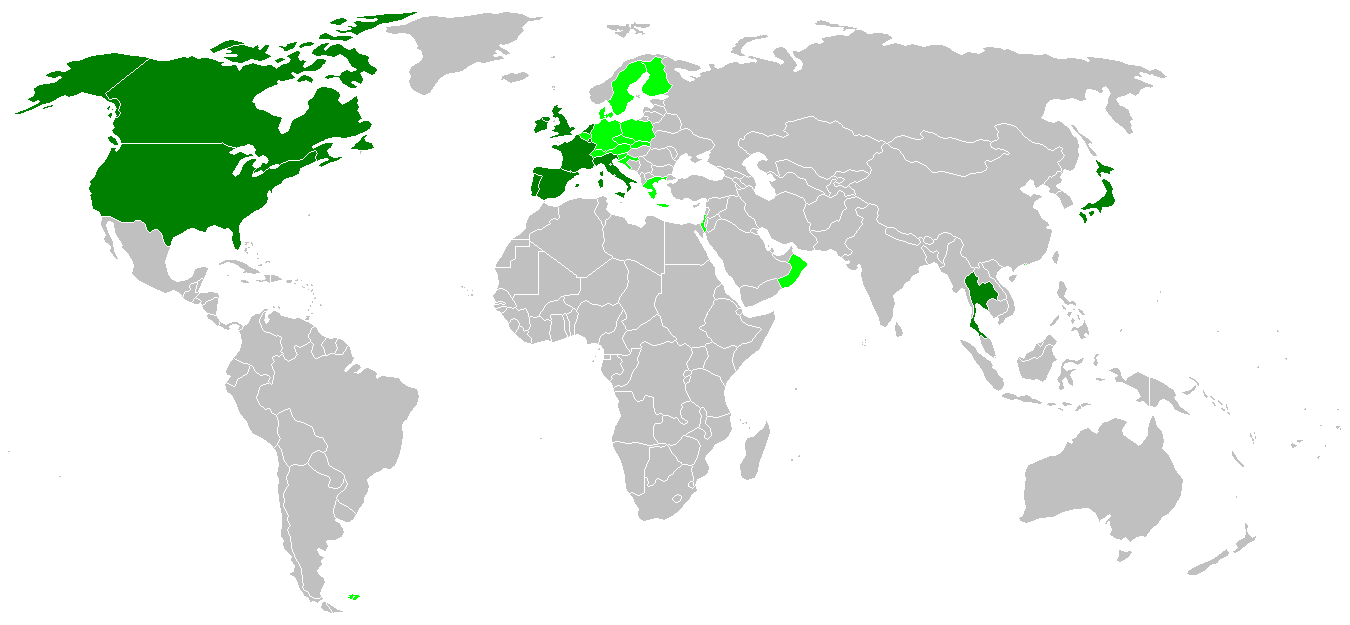
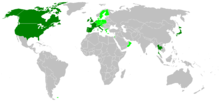 Dark green areas are countries that have confirmed human cases of variant Creutzfeldt-Jakob disease and light green are countries that have bovine spongiform encephalopathy cases.
Dark green areas are countries that have confirmed human cases of variant Creutzfeldt-Jakob disease and light green are countries that have bovine spongiform encephalopathy cases.Although CJD is the most common human prion disease, it is still rare, occurring in about one out of every one million people every year. It usually affects people aged 45–75, most commonly appearing in people between the ages of 60–65. The exception to this is the more recently-recognised 'variant' CJD (vCJD), which occurs in younger people.
CDC monitors the occurrence of CJD in the United States through periodic reviews of national mortality data. According to the CDC:
- CJD occurs worldwide at a rate of about 1 case per million population per year.
- On the basis of mortality surveillance from 1979 to 1994, the annual incidence of CJD remained stable at approximately 1 case per million persons in the United States.
- In the United States, CJD deaths among persons younger than 30 years of age are extremely rare (fewer than five deaths per billion per year[citation needed]).
- The disease is found most frequently in patients 55–65 years of age, but cases can occur in people older than 90 years and younger than 55 years of age.
- In more than 85% of cases, the duration of CJD is less than 1 year (median: four months) after onset of symptoms.[2][44]
New concerns
In The Lancet (June 2006), a University College London team suggested that it may take more than 50 years for vCJD to develop, from their studies of kuru, a similar disease in Papua New Guinea.[45] The reasoning behind the claim is that kuru was possibly transmitted through cannibalism in Papua New Guinea when family members would eat the body of a dead relative as a sign of mourning. In the 1950s, cannibalism was banned. In the late 20th century, however, kuru reached epidemic proportions in certain Papua New Guinean communities, therefore suggesting that vCJD may also have a similar incubation period of 30 to 50 years. A critique to this theory is that while mortuary cannibalism was banned in Papua New Guinea in the 1950s, that does not necessarily mean that the practice ended. 15 years later Jared Diamond was informed by Papuans that the practice continued.[46] Kuru may have passed to the Fore people through the preparation of the dead body for burial.
These researchers noticed a genetic variation in some kuru patients that has been known to promote long incubation periods. They have also proposed that individuals who contracted CJD in the early 1990s represent a distinct genetic subpopulation, with unusually short incubation periods for Bovine spongiform encephalopathy (BSE). This means that there may be many more vCJD patients who have longer incubation periods, which may surface many years later.[45]
In 1997 a number of Kentuckians developed CJD. It was discovered that all the victims had consumed squirrel brains, although a coincidental relationship between the disease and this dietary practice may have been involved.[47] See: http://www.guardian.co.uk/uk/2008/aug/03/bse.medicalresearch for recent concerns.
History
The disease was first described by German neurologist Hans Gerhard Creutzfeldt in 1920 and shortly afterwards by Alfons Maria Jakob, giving it the name Creutzfeldt–Jakob. Some of the clinical findings described in their first papers do not match current criteria for Creutzfeldt–Jakob disease, and it has been speculated that at least two of the patients in initial studies were suffering from a different ailment.[citation needed] An early description of familial CJD stems from the German psychiatrist and neurologist Friedrich Meggendorfer (1880–1953).[48][49]
References
- ^ Merriam-Webster's Collegiate Dictionary
- ^ a b "CJD (Creutzfeldt-Jakob Disease, Classic)". Centers for Disease Control and Prevention. 2008-02-26. http://www.cdc.gov/ncidod/dvrd/cjd/index.htm. Retrieved 2009-06-20.
- ^ Paul Brown (2001-07-04). "Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease". BMJ. http://www.bmj.com/content/322/7290/841.full. Retrieved 2011-02-23.
- ^ "UW Hospital warns 53 patients about possible exposure to fatal disease". Wisconsin State Journal. 2009-07-24. http://www.madison.com/wsj/topstories/459435. Retrieved 2009-07-24.[dead link]
- ^ Niimi Y, Iwasaki Y, Umemura T (December 2008). "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course". Neuropathology 28 (6): 645–51. doi:10.1111/j.1440-1789.2008.00904.x. PMID 18410280. http://www3.interscience.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0919-6544&date=2008&volume=28&issue=6&spage=645.
- ^ Jones M, Peden AH, Prowse CV (September 2007). "In vitro amplification and detection of variant Creutzfeldt–Jakob disease PrPSc". J. Pathol. 213 (1): 21–6. doi:10.1002/path.2204. PMID 17614097.
- ^ Frosh A, Joyce R, Johnson A (June 2001). "Iatrogenic vCJD from surgical instruments". BMJ 322 (7302): 1558–9. doi:10.1136/bmj.322.7302.1558. PMC 1120609. PMID 11431283. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1120609.
- ^ Will RG, Ironside JW, Zeidler M (April 1996). "A new variant of Creutzfeldt–Jakob disease in the UK". Lancet 347 (9006): 921–5. doi:10.1016/S0140-6736(96)91412-9. PMID 8598754.
- ^ Wang XF, Dong CF, Zhang J (March 2008). "Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro". Mol. Cell. Biochem. 310 (1–2): 49–55. doi:10.1007/s11010-007-9664-6. PMID 18038270.
- ^ a b Gambetti, Pierluigi. "Creutzfeldt-Jakob Disease (CJD)". The Merck Manuals: Online Medical Library. http://www.merckmanuals.com/home/sec06/ch090/ch090b.html. Retrieved 4/6/11.
- ^ Chakraborty C, Nandi S, Jana S (April 2005). "Prion disease: a deadly disease for protein misfolding". Current Pharmaceutical Biotechnology 6 (2): 167–77. doi:10.2174/1389201053642321. PMID 15853695.
- ^ a b Obi RK, Nwanebu FC (2008). "Prions And Prion Diseases". African Journal of Clinical and Experimental Microbiology 9 (1): 38–52. ISSN 1595-689X. http://ajol.info/index.php/ajcem/article/view/7481. Retrieved 2009-06-20.
- ^ Manuelidis L, Yu ZX, Barquero N, Banquero N, Mullins B (February 2007). "Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles". Proceedings of the National Academy of Sciences of the United States of America 104 (6): 1965–70. doi:10.1073/pnas.0610999104. PMC 1794316. PMID 17267596. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1794316. Retrieved 2009-06-20.
- ^ Mills JL, Schonberger LB, Wysowski DK (April 2004). "Long-term mortality in the United States cohort of pituitary-derived growth hormone recipients". The Journal of Pediatrics 144 (4): 430–6. doi:10.1016/j.jpeds.2003.12.036. PMID 15069388. Lay summary – Wired (2004-04-09).
- ^ "Questions and Answers: Creutzfeldt–Jakob Disease Infection-Control Practices". Infection Control Practices/CJD (Creutzfeldt–Jakob Disease, Classic). Centers for Disease Control and Prevention. January 4, 2007. http://www.cdc.gov/ncidod/dvrd/cjd/qa_cjd_infection_control.htm#sterilization. Retrieved 2007-06-09.
- ^ "WHO Infection Control Guidelines for Transmissible Spongiform Encephalopathies". World Health Organization: Communicable Disease Surveillance and Control. 26 March 1999. http://www.who.int/csr/resources/publications/bse/WHO_CDS_CSR_APH_2000_3/en/. Retrieved 2007-06-09.
- ^ McDonnell G, Burke P (May 2003). "The challenge of prion decontamination". Clinical Infectious Diseases 36 (9): 1152–4. doi:10.1086/374668. PMID 12715310.
- ^ Solassol J, Pastore M, Crozet C (2006). "A novel copper–hydrogen peroxide formulation for prion decontamination". J Infect Dis 194 (6): 865–869. doi:10.1086/506947. PMID 16941355.
- ^ Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW (2004). "Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient". Lancet 364 (9433): 527–9. doi:10.1016/S0140-6736(04)16811-6. PMID 15302196.
- ^ "Variant CJD and blood donation" (PDF). National Blood Service. August 2004. http://www.blood.co.uk/pdfdocs/vcjd.pdf. Retrieved 2009-06-20.[dead link]
- ^ Regan F, Taylor C (July 2002). "Blood transfusion medicine". BMJ (Clinical Research Ed.) 325 (7356): 143–7. doi:10.1136/bmj.325.7356.143. PMC 1123672. PMID 12130612. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1123672. Retrieved 2009-06-20.
- ^ "In-Depth Discussion of Variant Creutzfeld-Jacob Disease and Blood Donation". American Red Cross. Archived from the original on 2007-12-30. http://web.archive.org/web/20071230205118/http://www.redcross.org/services/biomed/blood/supply/cjdv.html. Retrieved 2009-06-20.
- ^ "Permanent exclusion criteria" (in German). Blutspendedienst Hamburg. http://www.blutspendehamburg.de/blutspender-gesucht/ausschluss. Retrieved 2009-06-20.
- ^ "Donor Qualification". Héma-Québec. 2009-03-26. http://www.hema-quebec.qc.ca/anglais/dondesang/qualifidonneurs.htm. Retrieved 2009-06-20.[dead link]
- ^ "Permanent exclusion criteria / Dyskwalifikacja stała" (in Polish). RCKiK Warszawa. http://www.rckik-warszawa.com.pl/dlakrwio_ds.html. Retrieved 2010-03-03.[dead link]
- ^ "Blood donor guidance / Poučení dárce krve" (in Czech). Fakultní nemocnice Královské Vinohrady. http://www.fnkv.cz/soubory/pouceni-darce-krve-2007.doc. Retrieved 2010-03-20.
- ^ Stein, Rob (August 13, 2008). "Mad Cow Rules Hit Sperm Banks' Patrons". washingtonpost.com (The Washington Post Company). http://www.washingtonpost.com/wp-dyn/content/article/2008/08/12/AR2008081203131.html. Retrieved 2008-10-04.
- ^ a b Kotler, Steven (2007-09-27). "The God of Sperm". LA Weekly. http://www.laweekly.com/2007-09-27/news/the-god-of-sperm/. Retrieved 2009-06-20.
- ^ By Rachael Rettner (2011-02-03). "Blood test may screen for human form of mad cow". MSNBC. http://www.msnbc.msn.com/id/41406612/ns/health-infectious_diseases/. Retrieved 2011-02-09.
- ^ Young, Geoffrey S.; G; F; M; H; L; L; W et al. (June–July 2005). "Diffusion-Weighted and Fluid-Attenuated Inversion Recovery Imaging in Creutzfeldt–Jakob Disease: High Sensitivity and Specificity for Diagnosis". American Journal of Neuroradiology (American Society of Neuroradiology) 26 (6): 1551–1562. PMID 15956529. http://www.ajnr.org/cgi/content/full/26/6/1551. Retrieved 2007-10-30.
- ^ Tschampa, Henriette J.; M; F; P; S; U (1 May 2003). "Thalamic Involvement in Sporadic Creutzfeldt–Jakob Disease: A Diffusion-Weighted MR Imaging Study". American Journal of Neuroradiology (American Society of Neuroradiology) 24 (5): 908–915. PMID 12748093. http://www.ajnr.org/cgi/content/full/24/5/908. Retrieved 2007-10-30.
- ^ Sanchez-Juan, P., Green, A., Ladogana, A., et al. (2006). "CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease". Neurology 67 (4): 637–643. doi:10.1212/01.wnl.0000230159.67128.00. PMID 16924018.
- ^ Tattum, M. H., Jones, S., Pal, S., Khalili-Shirazi, A., Collinge, J., Jackson, G. (December 2010). "A highly sensitive immunoassay for the detection of prion-infected material in whole human blood without the use of proteinase K". Transfusion (AABB) 50: 2619–2627. doi:10.1111/j.1537-2995.2010.02731.x.
- ^ "Detecting Prions in Blood". Microbiology Today: 195. August 2010. https://www.sgm.ac.uk/pubs/micro_today/pdf/081010.pdf. Retrieved 2011-08-21.
- ^ "SOFIA: An Assay Platform for Ultrasensitive Detection of PrPSc in Brain and Blood". SUNY Downstate Medical Center. http://www.bionosis.com/news/Bionosis%20PrioNet%20Poster.pdf. Retrieved 2011-08-19.
- ^ a b Sternberg's Diagnostic Surgical Pathology, 5th edition.
- ^ http://library.med.utah.edu/WebPath/TUTORIAL/CNS/CNSDG.html
- ^ Belay ED, Schonberger LB (2002). "Variant Creutzfeldt–Jakob disease and bovine spongiform encephalopathy". Clin. Lab. Med. 22 (4): 849–62, v–vi. doi:10.1016/S0272-2712(02)00024-0. PMID 12489284.
- ^ "Teenager with vCJD 'stable". London: BBC News. 13 December 2004. http://news.bbc.co.uk/2/hi/uk_news/northern_ireland/4092363.stm. Retrieved 2007-01-01.
- ^ Bone, Ian (12 July 2006). "Intraventricular Pentosan Polysulphate in Human Prion Diseases: A study of Experience in the United Kingdom". Medical Research Council. http://www.mrc.ac.uk/NewsViewsAndEvents/News/MRC001707. Retrieved 2007-01-01.
- ^ "Use of Pentosan Polysulphate in the treatment of, or prevention of, vCJD". Department of Health:CJD Therapy Advisory Group. http://www.dh.gov.uk/en/Policyandguidance/Healthandsocialcaretopics/CJD/CJDgeneralinformation/DH_4031039. Retrieved 2007-10-30.
- ^ Rainov NG, Tsuboi Y, Krolak-Salmon P, Vighetto A, Doh-Ura K (2007). "Experimental treatments for human transmissible spongiform encephalopathies: is there a role for pentosan polysulfate?". Expert opinion on biological therapy 7 (5): 713–26. doi:10.1517/14712598.7.5.713. PMID 17477808.
- ^ Pfeifer A, Eigenbrod S, Al-Khadra S (December 2006). "Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice". The Journal of Clinical Investigation 116 (12): 3204–10. doi:10.1172/JCI29236. PMC 1679709. PMID 17143329. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1679709. Retrieved 2009-06-20. Lay summary – BBC News (2006-12-04).
- ^ "vCJD (Variant Creutzfeldt-Jakob Disease)". Centers for Disease Control and Prevention. 2007-01-04. http://www.cdc.gov/ncidod/dvrd/vcjd/factsheet_nvcjd.htm. Retrieved 2009-06-20.
- ^ a b Collinge J, Whitfield J, McKintosh E (June 2006). "Kuru in the 21st century--an acquired human prion disease with very long incubation periods". Lancet 367 (9528): 2068–74. doi:10.1016/S0140-6736(06)68930-7. PMID 16798390.
- ^ Diamond, JM (7 September 2000). "Archaeology: Talk of cannibalism". Nature 407 (25-26): 25. doi:10.1038/35024175.
- ^ Berger JR, Waisman E, Weisman B (August 1997). "Creutzfeldt-Jakob disease and eating squirrel brains". Lancet 350 (9078): 642. doi:10.1016/S0140-6736(05)63333-8. PMID 9288058.
- ^ Meggendorfer F. Klinische und genealogische Beobachtungen bei einem Fall von spastischer Pseudokosklerose Jakobs. Z Neurol Psychiatry 1930; 128: 337–41
- ^ Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003). "Sporadic and familial CJD: classification and characterisation". Br. Med. Bull. 66: 213–39. PMID 14522861.
External links
- UCSF Memory and Aging Center - education website from a CJD patient care and research center
- UCSF Memory and Aging Center YouTube channel with films about CJD
- Institute for Neurodegenerative Diseases (IND) - a basic science research institute working on a cure for CJD
- Creutzfeldt–Jakob disease at the Open Directory Project
Infectious diseases – Prion diseases / Transmissible spongiform encephalopathy (A81, 046) Prion diseases in humans sporadic: sCJDacquired/transmissible: Kuru · vCJDPrion diseases in animals Bovine spongiform encephalopathy · Scrapie · Chronic wasting disease · Transmissible mink encephalopathy · Feline spongiform encephalopathy · Exotic ungulate encephalopathyConsumer food safety Adulterants/food contaminants 3-MCPD · Aldicarb · Cyanide · Formaldehyde · Lead poisoning · Melamine · Mercury in fish · Sudan red dye
Toxins/poisons Aflatoxin · Arsenic contamination of groundwater · Benzene in soft drinks · Bisphenol A · Mycotoxins · Shellfish poisoning
Microorganisms Pesticides overuse/residues Chlorpyrifos · DDT · Lindane · Malathion · Methamidophos
Preservatives Sweeteners Acesulfame potassium · Aspartame · Cyclamate · High-fructose corn syrup (Health effects, Public relations) · Saccharin · Sorbitol · Sucralose
Food scares 2005 Indonesia food scare · 2006 North American E. coli outbreak · 2007 Vietnam food scare · 2008 Canadian listeriosis outbreak · 2008 Chinese milk scandal · 2008 Irish pork crisis · 2008 United States salmonellosis outbreak · 2011 E. coli O104:H4 outbreak · Food safety incidents in the People's Republic of China · Bradford sweets poisoning · Chilean grape scare · ICA meat repackaging controversy · Minamata disease · Toxic oil syndrome · List of foodborne illness outbreaks · List of food contamination incidents
Regulatory/watchdog Acceptable daily intake · E number · Early history of food regulation in the United States) · European Food Safety Authority · Food and Drug Administration · Food labeling regulations · (United Kingdom and United States Food law · Food Safety Act 1990 · Food Standards Agency · International Food Safety Network · Pure Food and Drug Act · Quality Assurance International · List of food safety organisations
Food processing 4-Hydroxynonenal · Acrylamide · Creutzfeldt–Jakob disease · Food irradiation · Heterocyclic amines · Hydrolyzed vegetable protein · Modified starch · Nitrosamines · Polycyclic aromatic hydrocarbon · Shortening · Trans fat
Miscellaneous Categories:- Transmissible spongiform encephalopathies
- Neurodegenerative disorders
- Viral infections of the central nervous system
- Rare diseases

Wikimedia Foundation. 2010.