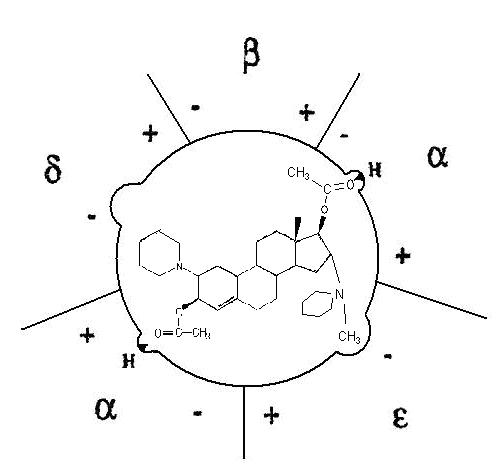
- Neuromuscular-blocking drug
-
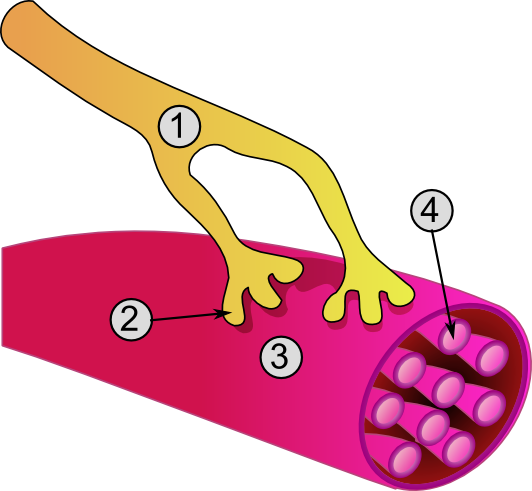
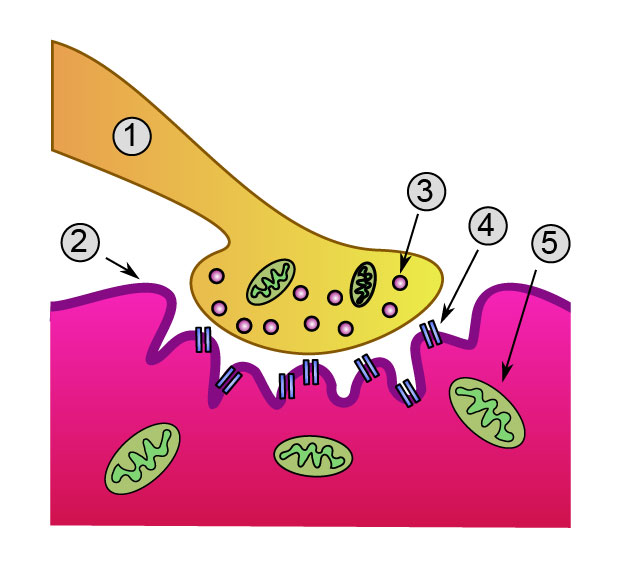
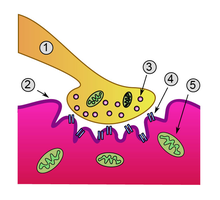 Detailed view of a neuromuscular junction:
Detailed view of a neuromuscular junction:
1. Presynaptic terminal
2. Sarcolemma
3. Synaptic vesicle
4. Nicotinic acetylcholine receptor
5. MitochondrionNeuromuscular blocking drugs block neuromuscular transmission at the neuromuscular junction,[1] causing paralysis of the affected skeletal muscles. This is accomplished either by acting presynaptically via the inhibition of acetylcholine (ACh) synthesis or release, or by acting postsynaptically at the acetylcholine receptors of the motor nerve end-plate. While there are drugs that act presynaptically (such as botulinum toxin and tetanus toxin), the clinically-relevant drugs work postsynaptically.
Clinically, neuromuscular block is used adjunctively to anesthesia to produce paralysis, so that surgery, especially intra-abdominal and intra-thoracic surgeries, can be conducted with fewer complications. Because the appropriate dose of neuromuscular blocking drug may paralyze muscles required for breathing (i.e. the diaphragm), mechanical ventilation should be available to maintain adequate respiration.
Patients are still aware of pain even after full conduction block has occurred; hence, general anesthetics and/or analgesics must also be given to prevent anesthesia awareness.
Quaternary ammonium muscle relaxants are quaternary ammonium salts used as drugs for muscle relaxation, most commonly in anesthesia. It is necessary to prevent spontaneous movement of muscle during surgical operations. Muscle relaxants inhibit neuron transmission to muscle by blocking the nicotinic acetylcholine receptor. What they have in common, and is necessary for their effect, is the structural presence of quaternary ammonium groups, usually two. Some of them are found in nature and others are synthesized molecules.[2][3]
Contents
History
Curare is a crude extract from South American plants. It was known in the 19th century to have a paralysing effect. D-tubocurarine a mono-quaternary alkaloid was isolated from Chondodendron tomentosum in 1942, and it was shown to be the active chemical in curare that had the paralysing effect. At that time it was known that curare and therefore d-tubocurarine worked at the neuromuscular junction. The isolation of tubocurarine and its marketing as the drug Intocostrin led to more research in the field of neuromuscular blocking drugs. Scientists figured out that the potency of tubocurarine was related to the separation distance between the two quaternary ammonium heads.[2][4]
Further research led to the development of synthesized molecules with different curariform effects, depending on the distance between the quaternary ammonium groups. One of the synthesized bis-quaternaries was decamethonium a 10-carbon bis-quaternary compound. Following research with decamethonium, scientists developed suxamethonium, which is a double acetylcholine molecule that was connected at the acetyl end. The discovery and development of suxamethonium lead to a Nobel Prize in medicine in 1957. Suxamethonium showed different blocking effect in that its effect was achieved more quickly and augmented a response in the muscle before block. Also, tubocurarine effects were known to be reversible by acetylcholinesterase inhibitors, whereas decamethonium and suxamethonium block were not reversible.[2][3]
Another compound malouétine that was a bis-quaternary steroid was isolated from the plant Malouetia bequaertiana and showed curariform activity. This led to the synthetic drug pancuronium a bis-quaternary steroid and subsequently other chemicals that had better pharmacological properties as drugs.[2][5]
These molecules discussed above and research devoted to them led to many other compounds with different pharmacological properties and research in drug development as well as they helped to better the understanding the physiology of neurons and receptors.
Nomenclature
Neuromuscular blocking drugs are often classified into two broad classes: Pachycurares which are bulky molecules with nondepolarizing activity. Leptocurares are on the other hand thin and flexible molecules. They tend to have depolarizing activity.[3] It is also common to classify them based on their chemical structure.
- Acetylcholine, succinylcholine, and decamethonium
Succinylcholine was synthesised by connecting two acetylcholine molecules and has the same number of heavy atoms between methonium heads as decamethonium. Just like acetylcholine, succinylcholine, decamethonium and other polymethylene chains, of the appropriate length and with two methonium, heads have small trimethyl onium heads and flexible links. They all exhibit a depolarizing block.
- Aminosteroids
Pancuronium, vecuronium, rocuronium, rapacuronium, dacuronium, malouètine, duador, dipyrandium, pipecuronium, chandonium (HS-310), HS-342 and other HS- compounds are aminosteroidal agents. They have in common the steroid structural base which provides a rigid and bulky body. Most of the agents in this category would also be classified as non-depolarizing.
- Tetrahydroisoquinoline derivatives
Compounds based on the tetrahydroisoquinoline moiety such as atracurium, mivacurium, and doxacurium would fall in this category. They have a long and flexible chain between the onium heads, except for the double bond of mivacurium. D-tubocurarine and dimethyltubocurarine are also in this category. Most of the agents in this category would be classified as non-depolarizing.
- Gallamine and other chemical classes
Gallamine is a trisquaternary ether with three ethonium heads attached to a phenyl ring through an ether linkage. Many other different structures have been used for their muscle relaxant effect such as alcuronium (alloferin), anatruxonium, diadonium, fazadinium (AH8165) and tropeinium.
- Novel NMB agents
In recent years much research has been devoted to new types of quaternary ammonium muscle relaxants. These are asymmetrical diester isoquinolinium compounds and bis-benzyltropinium compounds that are bistropinium salts of various diacids. These classes have been developed to create muscle relaxants that are faster and shorter acting. Both the asymmetric structure of diester isoquinolinium compounds and the acyloxylated benzyl groups on the bisbenzyltropiniums destabilizes them and can lead to spontaneous breakdown and therefore possibly a shorter duration of action.[6]
Classification
These drugs fall into two groups:
- Non-depolarizing blocking agents: These agents constitute the majority of the clinically-relevant neuromuscular blockers. They act by competitively blocking the binding of ACh to its receptors, and in some cases, they also directly block the ionotropic activity of the ACh receptors.[7]
- Depolarizing blocking agents: These agents act by depolarizing the plasma membrane of the skeletal muscle fiber. This persistent depolarization makes the muscle fiber resistant to further stimulation by ACh.
Non-depolarizing blocking agents
A neuromuscular nondepolarizing agent is a form of neuromuscular blocker which do not depolarize the motor end plate.[8]
The quaternary ammonium muscle relaxants belong to this class.
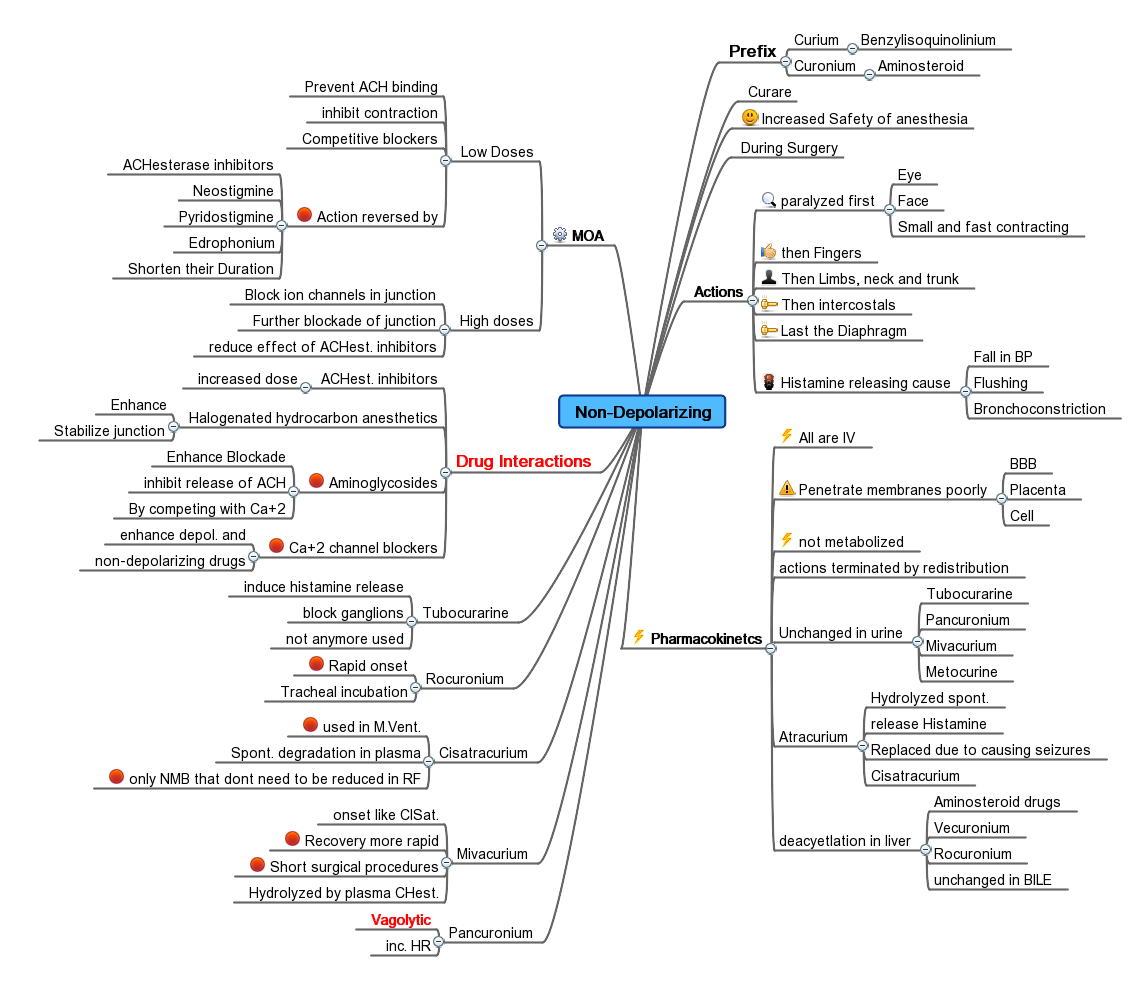
 Mind Map showing a summary of Neuromuscular nondepolarizing agent
Mind Map showing a summary of Neuromuscular nondepolarizing agentBelow are some of the more common agents that act as competitive antagonists against acetylcholine at the site of postsynaptic acetylcholine receptors.
Tubocurarine, found in curare of the South American plant Pareira, Chonodendron tomentosum, is the prototypical non-depolarizing neuromuscular blocker. It has a slow onset (>5 min) and a long duration of action (1–2 hours). Side effects include hypotension, which is partially explained by its effect of increasing histamine release, a vasodilator,[9] as well as its effect of blocking autonomic ganglia.[10] It is excreted in the urine.
This drug needs to block about 70-80% of the Ach receptors for neuromuscular conduction to fail, and hence, for effective blockade to occur. At this stage, end-plate potentials (EPPs) can still be detected, but are too small to reach the threshold potential needed for activation of muscle fiber contraction.
Comparison of non-depolarizing neuromuscular blocking agents Agent Time to onset
(seconds)Duration
(minutes)Side-effects Clinical use Storage Rapacuronium (Raplon) Mivacurium (Mivacron) 90 12-18[11] - hypotension (transiently), by release of histamine[11]
No longer manufactured secondary to marketing, manufacturing, financial concerns refrigerated Atracurium (Tracrium) 90 30 min or less[11] - hypotension, transiently,[11] by release of histamine
- Toxic metabolite called laudanosine, greater accumulation in individuals with renal failure
widely[11] refrigerated Doxacurium (Nuromax) long[11] - hypotension, transiently,[11] by release of histamine
- Toxic metabolite called laudanosine, greater accumulation in individuals with renal failure
Cisatracurium (Nimbex) 90 60-80 does not cause release of histamine refrigerated Vecuronium (Norcuron) 60 30-40[11] Few,[11] may cause prolonged paralysis[11] and promote muscarinic block widely[11] non-refrigerated Rocuronium (Zemuron) 75 45-70[citation needed] may promote muscarinic block non-refrigerated Pancuronium (Pavulon) 90 180 or more[citation needed] - tachycardia (slight)[11]
(no hypotension)[11]
widely[11] non-refrigerated Tubocurarine (Jexin) 300 or more[11] 60 - 120[11] - hypotension (by ganglion-block and histamine release)[11]
- Bronchoconstriction (by histamine release)[11]
rarely[11] gallamine (Flaxedil) 300 or more[11] 60 - 120[11] Pipecuronium 90 180 or more[citation needed] - tachycardia (slight)[11]
(no hypotension)[11]
non-refrigerated Depolarizing blocking agents
A neuromuscular depolarizing blocking agent is a form of neuromuscular blocker which will depolarize the motor end plate.[12]
An example is succinylcholine.
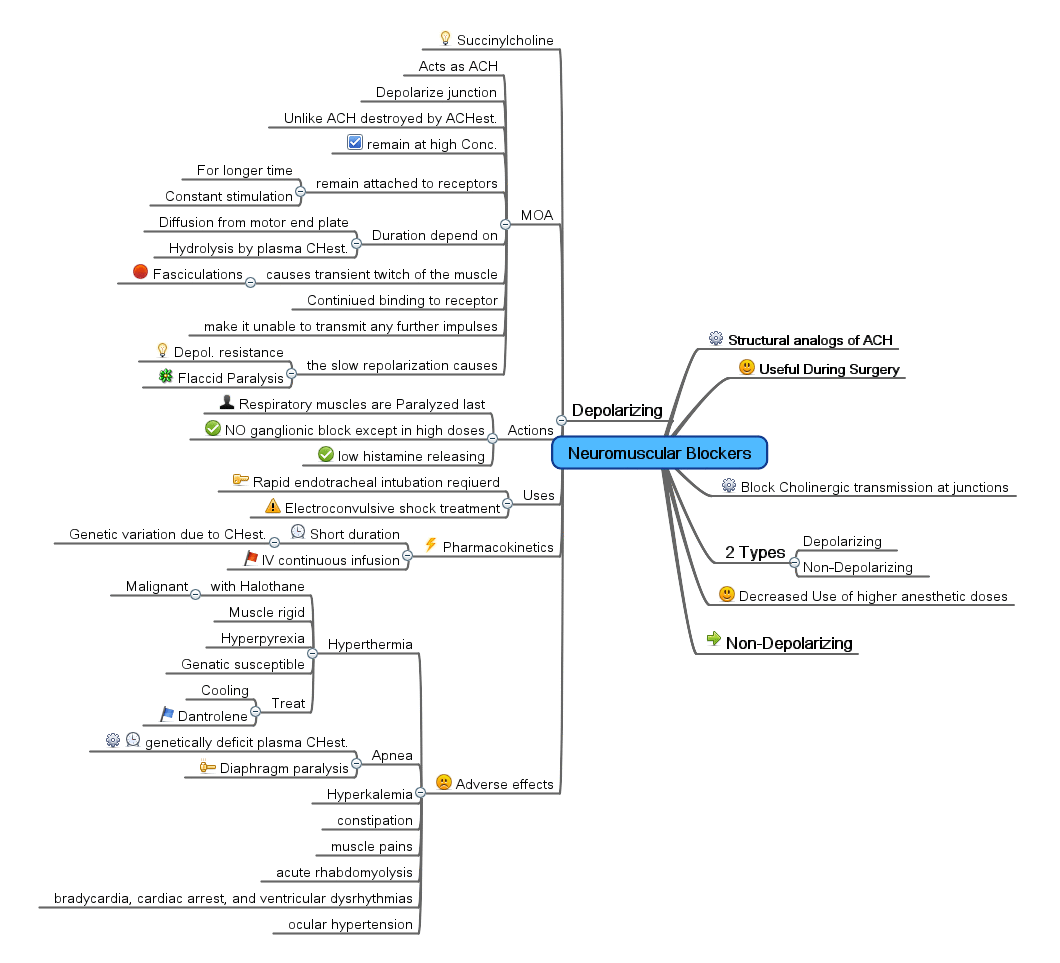
 Mind Map showing a summary of Neuromuscular depolarizing agent
Mind Map showing a summary of Neuromuscular depolarizing agentDepolarizing blocking agents work by depolarizing the plasma membrane of the muscle fiber, similar to acetylcholine. However, these agents are more resistant to degradation by acetylcholinesterase, the enzyme responsible for degrading acetylcholine, and can thus more persistently depolarize the muscle fibers. This differs from acetylcholine, which is rapidly degraded and only transiently depolarizes the muscle.
There are two phases to the depolarizing block. During phase I (depolarizing phase), they cause muscular fasciculations (muscle twitches) while they are depolarizing the muscle fibers. Eventually, after sufficient depolarization has occurred, phase II (desensitizing phase) sets in and the muscle is no longer responsive to acetylcholine released by the motoneurons. At this point, full neuromuscular block has been achieved.
The prototypical depolarizing blocking drug is succinylcholine (suxamethonium). It is the only such drug used clinically. It has a rapid onset (30 seconds) but very short duration of action (5–10 minutes) because of hydrolysis by various cholinesterases (such as butyrylcholinesterase in the blood). Succinylcholine was originally known as diacetylcholine because structurally it is composed of two acetylcholine molecules joined with a methyl group. Decamethonium is sometimes, but rarely, used in clinical practice.
Comparison of drugs
The main difference is in the reversal of these two types of neuromuscular-blocking drugs.
- Non-depolarizing blockers are reversed by acetylcholinesterase inhibitor drugs since they are competitive antagonists at the ACh receptor so can be reversed by increases in ACh.
- The depolarizing blockers already have ACh-like actions, so these agents will have prolonged effect under the influence of acetylcholinesterase inhibitors. The administration of depolarizing blockers will initially exhibit fasciculations (a sudden twitch just before paralysis occurs). This is due to the depolarization of the muscle. Also, post-operative pain is associated with depolarizing blockers.
The tetanic fade is the failure of muscles to maintain a fused tetany at sufficiently-high frequencies of electrical stimulation.
- Non-depolarizing blockers will have this effect on patients, probably by an effect on presynaptic receptors.[13]
- Depolarizing blockers will not.
This discrepancy is diagnostically useful in case of intoxication of an unknown neuromuscular-blocking drug.[13]
Mechanism of action
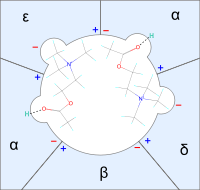 Fig.1 A simple illustration of how two acetylcholine molecules bind to its receptive sites on the nicotinic receptor
Fig.1 A simple illustration of how two acetylcholine molecules bind to its receptive sites on the nicotinic receptorQuaternary muscle relaxants bind to the nicotinic acetylcholine receptor and inhibit or interfere with the binding and effect of ACh to the receptor. Each ACh-receptor has two receptive sites and activation of the receptor requires binding to both of them. Each receptor site is located at one of the two α-subunits of the receptor. Each receptive site has two subsites, an anionic site that binds to the cationic ammonium head and a site that binds to the blocking agent by donating a hydrogen bond.[3]
Non-depolarizing agents A decrease in binding of acetylcholine leads to a decrease in its effect and neuron transmission to the muscle is less likely to occur. It is common understanding that non depolarizing agents block by acting as reversible competitive inhibitors. That is they bind to the receptor as antagonists and that leaves fewer receptors available for acetylcholine to bind to.[3][14]
Depolarizing agents Depolarizing agents produce their block by binding to and activating the receptor, causing contraction and after that paralysation. They bind to the receptor and cause depolarization by opening channels just like acetylcholine does. This causes repetitive excitation that lasts longer than a normal acetylcholine excitation and is most likely explained by depolarizing agents resistance to the enzyme acetylcholine esterase. The constant depolarization and triggering of the receptors keeps the endplate resistant to activation by acetylcholine. Therefor a normal neuron transmission to muscle can not cause contraction of the muscle because the endplate is depolarized and thereby the muscle paralysed.[3][14]
Binding to the nicotinic receptor Shorter molecules like acetylcholine need two molecules to activate the receptor, one at each receptive site. Decamethonium congeners, which prefer straight line conformations (their lowest energy state), will likely span the two receptive sites with one molecule (binding inter-site). Longer congeners will be required to bend when fitting the receptive sites.
The more energy a molecule needs to bend and fit will usually result in lower potency.[15]
Structural and conformational action relationship
Conformational study on neuromuscular blocking drugs is relatively new and developing. Traditional SAR studies do not specify environmental factors on molecules. Computer based conformational searches assume the molecules to be in vacuo which is not the case in vivo. Solvation models take into account the effect of a solvent on the conformation of the molecule. However, no system of solvation can mimic the effect of the complex fluid composition of the body [16]
The division of muscle relaxants to rigid and non-rigid is at most qualitative. The energy required for conformational changes may give a more precise and quantitative picture. Energy required for reducing onium head distance in the longer muscle relaxant chains may quantify their ability to bend and fit its receptive sites [15] Using computers it is possible to calculate the lowest energy state conformer and thus most populated and best representing the molecule. This state is referred to as the global minimum. The global minimum for some simple molecules can be discovered quite easily with certainty. Such as for decamethonium the straight line conformer is clearly the lowest energy state. Some molecules on the other hand have many rotatable bonds and their global minimum can only be approximated[16]
Molecular length and rigidity
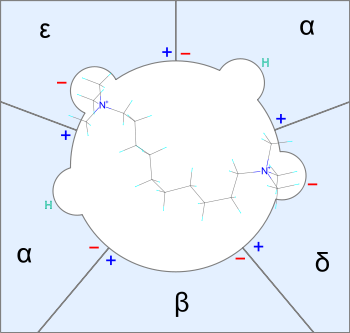 Fig.2 A simple illustration of how decamethonium binds to the nicotinic receptor. The onium heads bind to two separate subunits of the ion-channel
Fig.2 A simple illustration of how decamethonium binds to the nicotinic receptor. The onium heads bind to two separate subunits of the ion-channelNeuromuscular blocking agents need to fit in a space close to 2 nanometres which resembles the molecular length of decamethonium.[15] Some molecules of decamtehonium congeners may bind only to one receptive site. Flexible molecules will have a greater chance of fitting receptive sites. However the most populated conformation may not be the best fitted one. Very flexible molecules are in fact weak neuromuscular inhibitors with flat dose-response curves. On the other hand stiff or rigid molecules tend to fit well or not at all. If the lowest energy conformation fits the compound will have high potency because there will be a great concentration of molecules close to the lowest energy conformation. Molecules can be thin but yet rigid.[16] Decamethonium for example needs relatively high energy to change the N-N distance.[15]
Generally molecular rigidity contributes to potency while size affects whether a muscle relaxant will show a polarizing or a depolarizing effect.[6] Cations must be able to flow through the trans-membrane tube of the ion-channel to depolarize the endplate.[16] Small molecules may be rigid and potent but unable to occupy or block the area between the receptive sites.[6] Large molecules on the other hand may bind to both receptive sites and hinder depolarizing cations independent of whether the ion-channel is open or closed below. Having a lipophilic surface pointed towards the synapse will enhance this effect by repelling cations. The importance of this effect varies between different muscle relaxants and classifying depolarizing from non-depolarizing blocks is a complex issue. The onium heads are usually kept small and the chains connecting the heads usually keep the N-N distance at 10 N or O atoms. Keeping the distance in mind the structure of the chain can vary (double bonded, cyclohexyl, benzyl, etc.)[16]
Succinylcholine has a 10 atom distance between its N atoms, like decamethonium, but yet it has been reported that it takes two molecules, as with acetylcholine, to open one nicotinic ion channel. The conformational explanation for this would be that each acetylcholine moiety of succinylcholine prefers the gauche (bent, cis) state. The attraction between the N and O atoms is greater than the onium head repulsion. In this most populated state the N-N distance will be shorter than the optimal distance of ten carbon atoms and to short to occupy both receptive sites. This similarity between succinyl- and acetyl-choline also explains its acetylcholine-like side effects.[16] Comparing molecular lengths the pachycurares dimethyltubocurarine and d-tubocurarine which are both very rigid and both measure close to 1.8 nm in total length. Pancuronium and vecuronium measure 1.9 nm while pipecuronium is 2.1 nm. The potency of these compounds follows the same rank of order as their length. Likewise the leptocurares prefer a similar length. Decamethonium which measures 2 nm is the most potent in its category while C11 is slightly too long. Gallamine despite having low bulk and rigidity is the most potent in its class and it measures 1.9 nm.[6][15] Based on this information one can conclude that the optimum length for neuromuscular blocking agents, depolarizing or not, should be 2 to 2.1 nm.[16]
The CAR for long chain bisquaternary tetrahydroisoquinolines like atracurium, cisatracurium, mivacurium and doxacurium is hard to determine because of their bulky onium heads and large number of rotatable bonds and groups. These agents must follow the same receptive topology as others which means that they don’t fit between the receptive sites without bending.[15] Mivacurium for example has a molecular length of 3.6 nm when stretched out, far from the 2 to 2.1 nm optimum. Mivacurium, atracurium and doxacurium have greater N-N distance and molecular length than d-tubocurarine even when bent. To make them fit they have flexible connections that give their onium heads a chance to position themselves beneficially. This bent N-N scenario probably does not apply to laudexium and decamethylene bisatropium which prefer a straight conformation.[16]
Beers and Reich's law
It has been concluded that acetylcholine and related compounds must be in the gauche (bent) configuration when bound to the nicotinic receptor.[17]) Beers and Reich's studies on cholinergic receptors in 1970 showed a relationship affecting whether a compound was muscarinic or nicotinic. They showed that the distance from the centre of the quaternary N atom to the van der Waals extension of the respective O atom (or an equivalent H-bond acceptor) is a determining factor. If the distance is 0.44 nm then the compound will show muscarinic properties and if the distance is 0.59 nm then nicotinic properties will be dominant.[18])
Rational design
Pancuronium remains one of the few muscle relaxants logically and rationally designed from structure-action relationship data. A steroid skeleton was chosen because of its appropriate size and rigidness. Acetylcholine moieties were inserted to increase receptor affinity. Although having many unwanted side effects, a slow onset of action and recovery rate it was a big success and at the time the most potent neuromuscular drug available. Pancuronium and some other neuromuscular blocking agents block M2-receptors and therefore affect the vagus nerve leading to hypotension and tachycardia. This muscarinic blocking effect is related to the acetylcholine moiety on the A ring on pancuronium. Making the N atom on the A ring tertiary the ring loses its acetylcholine moiety and the resulting compound, vecuronium, has nearly 100 times less affinity to muscarin receptors while maintaining its nicotinic affinity and a similar duration of action. Vecuronium is therefore free from cardiovascular effects. [3] The D ring shows excellent properties validating Beers and Reich's rule with great precision. As a result vecuronium has the greatest potency and specificity of all mono-quaternary compounds.[16]
Potency
Two functional groups contribute significantly to aminosteroidal neuromuscular blocking potency, presumably to enable them to bind the receptor at two points. A bis-quaternary two point arrangement on A and D-ring (binding inter-site) or a D-ring acetylcholine moiety (binding at two points intra-site) are most likely to be successful. A third group can have variable effects.[16] The quaternary and acetyl groups on the A and D ring of pipecuronium prevent it from binding intra-site (binding to two points at the same site). Instead it must bind as bis-quaternary (inter-site).[6] These structures are very dissimilar from acetylcholine and free pipecuronium from nicotinic or muscarinic side effects linked to acetylcholine moiety. Also they protect the molecule from hydrolysis by cholinesterases, which explain its nature of kidney excretion. The four methyl-groups on the quaternary N atoms make it less lipophilic than most aminosteroids. This also affects pipecuroniums metabolism by resisting hepatic uptake, metabolism and biliary excretion. The length of the molecule (2.1 nm, close to ideal) and its rigidness make pipecuronium the most potent and clean one-bulk bis-quaternary. Even though the N-N distance (1.6 nm) is far away from what is considered ideal, its onium heads are well exposed and the quaternary groups help bringing together the onium heads to the anionic centers of the receptors without chirality issues.[16]
Adding more than two onium heads in general does not add to potency. Although the third onium head in gallamine seems to help position the two outside heads near the optimum molecular length, it can interfere unfavorably and gallamine turns out to be a weak muscle relaxant like all multi-quaternary compounds. Considering acetylcholine a quaternizing group larger than methyl and an acyl group larger than acetyl would reduce the molecules potency. The charged N and the carbonyl O atoms are distanced from structures they bind to on receptive sites and thus decreasing potency. The carbonyl O in vecuronium for example is thrusted outward to appose the H-bond donor of the receptive site. This also helps explain why gallamine, rocuronium and rapacuronium are of relatively low potency.[16] In general methyl quaternization is optimal for potency but opposing this rule the trimethyl derivatives of gallamine are of lower potency than gallamine. The reason for this is that gallamine has a suboptimal N-N distance. Substituting the ethyl groups with methyl groups would make the molecular length also shorter than optimal. Methoxylation of tetrahydroisoquinolinium agents seems to improve their potency. How methoxylation improves potency is still unclear. Histamine release is a common attribute of benzylisoquinolinium muscle relaxants. This problem generally decreases with increased potency and smaller doses. The need for larger doses increases the degree of this side effect. Conformational or structural explanations for histamine release are not clear.[16]
Pharmacokinetics
Metabolism and Hofmann elimination
 Fig.3 A simple illustration of how vecuronium binds to the nicotinic receptor. Its D-ring binds to the receptor at two points and the lipophillic side of the molecule repels cations from flowing through the ion-channel
Fig.3 A simple illustration of how vecuronium binds to the nicotinic receptor. Its D-ring binds to the receptor at two points and the lipophillic side of the molecule repels cations from flowing through the ion-channelDeacetylating vecuronium at position 3 results in a very active metabolite.[19] In the case of rapacuronium the 3-deacylated metabolite is even more potent than rapacuronium. As long as the D-ring acetylcholine moiety is unchanged they retain their muscle relaxing effect. Mono-quaternary aminosteroids produced with deacylation in position 17 on the other hand are generally weak muscle relaxants.[16] In the development of atracurium the main idea was to make use of Hofmann elimination of the muscle relaxant in vivo. When working with bisbenzyl-isoquinolinium types of molecules, inserting proper features into the molecule such as an appropriate electron withdrawing group then Hofmann elimination should occur at conditions in vivo. Atracurium, the resulting molecule breaks down spontaneously in the body to inactive compounds and being especially useful in patients with kidney or liver failure. Cis-atracurium is very similar to atracurium except it is more potent and has a weaker tendency to cause histamine release.[3]
Structure relations to onset time
The effect of structure on the onset of action is not very well known except that the time of onset seems to be inversely related to potency.[20] In general mono-quaternary aminosteroids are faster than bis-quaternary compounds which means they are also of lower potency. A possible explanation for this effect is that drug delivery and receptor binding are of a different timescale. Weaker muscle relaxants are given in larger doses so more molecules in the central compartment must diffuse into the effect compartment, which is the space within the mouth of the receptor, of the body. After delivery to the effect compartment then all molecules act quickly.[21] Therapeutically this relationship is very inconvenient because low potency, often meaning low specificity can decrease the safety margin thus increasing the chances of side effects. Additionally even though low potency usually accelerates onset of action it does not guaranty a fast onset. Gallamine for example is weak and slow. When fast onset is necessary then succinylcholine or rocuronium are usually preferable.[16]
Elimination
Muscle relaxants can have very different metabolic pathways and it is important that the drug does not accumulate if certain elimination pathways are not active, for example in kidney failure.
Adverse effects
Since these drugs may cause paralysis of the diaphragm, mechanical ventilation should be at hand to provide respiration.
Additionally, these drugs may exhibit cardiovascular effects, since they are not fully selective for the nicotinic receptor and hence may have effects on muscarinic receptors.[10] If nicotonic receptors of the autonomic ganglia or adrenal medulla are blocked, these drugs may cause autonomic symptoms. Additionally, neuromuscular blockers may facilitate histamine release, which causes hypotension, flushing, and tachycardia.
In depolarizing the musculature, suxamethonium may trigger a transient release of large amounts of potassium from muscle fibers. This puts the patient at risk for life-threatening complications, such as hyperkalemia and cardiac arrhythmias.
Certain drugs such as aminoglycoside antibiotics and polymyxin and some fluoroquinolones also have neuromuscular blocking action as their side effect.
Estimating effect
Methods for estimating the degree of neuromuscular block include valuation of muscular response to stimuli from surface electrodes, such as in the train-of-four test, wherein four such stimuli are given in rapid succession. With no neuromuscular blockade, the resultant muscle contractions will be of equal strength, but gradually decrease in case of neuromuscular blockade.[22] It is recommended during use of continuous-infusion neuromuscular blocking agents in intensive care.[23]
Reversal
The effect of non-depolarizing neuromuscular-blocking drugs may be reversed with anticholinesterases, with neostigmine and edrophonium as commonly used examples. Of these, edrophonium has a faster onset of action than neostigmine, but it is unreliable when used to antagonize deep neuromuscular block.[24] Anticholinesterases increase the amount of acetylcholine in the neuromuscular junction, so a prerequisite for their effect is that the neuromuscular block is not complete, because in case every acetylcholine receptor is blocked then it doesn't matter how much acetylcholine is present.
Sugammadex is a newer drug for reversing neuromuscular block by rocuronium in general anaesthesia. It is the first selective relaxant binding agent (SRBA).[25]
See also
References
- ^ "Dorlands Medical Dictionary:neuromuscular blocking agent". http://www.mercksource.com/pp/us/cns/cns_hl_dorlands_split.jsp?pg=/ppdocs/us/common/dorlands/dorland/one/000002250.htm.
- ^ a b c d T. Raghavendra (2002). "Neuromuscular blocking drugs: discovery and development". Journal of the Royal Society of Medicine 95 (7): 363–367. doi:10.1258/jrsm.95.7.363. PMC 1279945. PMID 12091515. http://www.jrsm.org/cgi/content/full/95/7/363.
- ^ a b c d e f g h W. C. Bowman (2006). "Neuromuscular block". British Journal of Pharmacology 147 (S1): S277–S286. doi:10.1038/sj.bjp.0706404. PMC 1760749. PMID 16402115. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1760749.
- ^ O.A.Nedergard, A et al. (2003). "Curare:Flying death". Pharmacology & Toxicology 92, 154–155..
- ^ A. G. McKenzie (2000). "Prelude to pancuronium and vecuronium". Anaesthesia 55 (6): 551–556. doi:10.1046/j.1365-2044.2000.01423.x. PMID 10866718.
- ^ a b c d e Lee, C. (2001). "Structure, conformation and action of neuromuscular blocking drugs". British Journal of Anaesthesia 87 (5): 755–769. doi:10.1093/bja/87.5.755. PMID 11878528. http://bja.oxfordjournals.org/cgi/content/abstract/87/5/755.
- ^ Bufler J, Wilhelm R, Parnas H, Franke C, Dudel J (1996). "Open channel and competitive block of the embryonic form of the nicotinic receptor of mouse myotubes by (+)-tubocurarine". J. Physiol. (Lond.) 495 ( Pt 1) (Pt 1): 83–95. PMC 1160726. PMID 8866353. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1160726.
- ^ MeSH Neuromuscular+Nondepolarizing+Agents
- ^ Inada E, Philbin DM, Machaj V, et al. (1986). "Histamine antagonists and d-tubocurarine-induced hypotension in cardiac surgical patients". Clin. Pharmacol. Ther. 40 (5): 575–80. doi:10.1038/clpt.1986.226. PMID 2429800.
- ^ a b Ostergaard D, Engbaek J, Viby-Mogensen J (1989). "Adverse reactions and interactions of the neuromuscular blocking drugs". Medical toxicology and adverse drug experience 4 (5): 351–68. PMID 2682131.
- ^ a b c d e f g h i j k l m n o p q r s t u v w Rang, H. P. (2003). Pharmacology. Edinburgh: Churchill Livingstone. ISBN 0-443-07145-4. OCLC 51622037. Page 151
- ^ MeSH Neuromuscular+Depolarizing+Agents
- ^ a b Rod Flower; Humphrey P. Rang; Maureen M. Dale; Ritter, James M. (2007). Rang & Dale's pharmacology. Edinburgh: Churchill Livingstone. ISBN 0-443-06911-5.
- ^ a b L.L Brunton, J.S.Lazo, K.L.Parker (2006). "Goodman & Gilman's The pahrmacological basis of Therapeutics". New York:McGraw-Hill, Inc.p 220-223..
- ^ a b c d e f Lee, C.; Jones, T. (2002). "Molecular conformation–activity relationship of decamethonium congeners". British Journal of Anaesthesia 88 (5): 692–699. doi:10.1093/bja/88.5.692. PMID 12067008. http://bja.oxfordjournals.org/cgi/content/abstract/88/5/692.
- ^ a b c d e f g h i j k l m n o Lee, Chingmuh (2003). "Conformation, action and mechanism of action of neuromuscular blocking muscle relaxants". Pharmacology and Therapeutics 98 (2): 143–169. doi:10.1016/S0163-7258(03)00030-5.
- ^ Spivak, C.E, J.A. Waters and R.S Aronstam (1 July 1989). "Binding of semirigid nicotinic agonists to nicotinic and muscarinic receptors". Molecular Pharmacology 36 (1): 177–184. PMID 2747625. http://molpharm.aspetjournals.org/cgi/content/abstract/36/1/177.
- ^ Beers, W.H and E. Reich (1970). "Structure and Activity of Acetylcholine". Nature 228 (5275): 917–922. doi:10.1038/228917a0.
- ^ Caldwell, J.E., J. Szenohradszky, V. Segredo, P.M. Wright, C. McLoughlin, M.L. Sharma, L.D. Gruenke, D.M. Fisher and R.D. Miller (1 September 1994). "The pharmacodynamics and pharmacokinetics of the metabolite 3- desacetylvecuronium (ORG 7268) and its parent compound, vecuronium, in human volunteers". American Society for Pharmacology and Experimental Therapeutics 270 (3): 1216–1222. PMID 7932174. http://jpet.aspetjournals.org/cgi/content/abstract/270/3/1216.
- ^ Kopman, Aaron F., Monica M. Klewicka, David J. Kopman and George G. Neuman (1999). "Molar potency is predictive of the speed of onset of neuromuscular block for agents of intermediate, short and ultrashort duration". Anesthesiology 90 (2): 425–431. doi:10.1097/00000542-199902000-00016. PMID 9952148.
- ^ Donati, F. and Claude Meistelman (2005). "A kinetic-dynamic model to explain the relationship between high potency and slow onset time for neuromuscular blocking drugs". Journal of pharmacokinetics and Pharmacodynamics 19 (5): 537–552. http://www.springerlink.com/content/j5527q19x6071106/.
- ^ thefreedictionary.com > train-of-four Citing: Mosby's Medical Dictionary, 8th edition.
- ^ Strange, C.; Vaughan, L.; Franklin, C.; Johnson, J. (1997). "Comparison of train-of-four and best clinical assessment during continuous paralysis". American journal of respiratory and critical care medicine 156 (5): 1556–1561. PMID 9372675.
- ^ Shorten, G. D.; Ali, H. H.; Goudsouzian, N. G. (1993). "Neostigmine and edrophonium antagonism of moderate neuromuscular block induced by pancuronium or tubocurarine". British journal of anaesthesia 70 (2): 160–162. doi:10.1093/bja/70.2.160. PMID 8435259.
- ^ Naguib M (2007). "Sugammadex: another milestone in clinical neuromuscular pharmacology". Anesth Analg 104 (3): 575–81. doi:10.1213/01.ane.0000244594.63318.fc. PMID 17312211.
External links
Skeletal muscle relaxants (M03) Peripherally acting
(primarily antinicotinic,
NMJ block)Curare alkaloidsultra-short duration: Gantacurium
short duration: Mivacurium • Chandonium
intermediate duration: Atracurium • Cisatracurium • Fazadinium • Rocuronium • Vecuronium
long duration: Doxacurium • Dimethyltubocurarine • Pancuronium • Pipecuronium • Laudexium • Gallamine
unsorted: Hexafluronium (Hexafluorenium)Choline derivatives: Suxamethonium (Succinylcholine)
Polyalkylene derivatives: HexamethoniumCentrally acting Carbamic acid estersBenzodiazepinesAnticholinergics (Antimuscarinics)OtherBaclofen • Chlormezanone • Chlorphenesin • Chlorzoxazone • Donepezil • Eperisone • Flopropione • Mephenesin • Mephenoxalone • Metaxalone • Phenyramidol • Pridinol • Promoxolane • Quinine • Thiocolchicoside • Tizanidine • Tolperisone • TrazodoneDirectly acting Cholinergics Receptor ligands Agonists: 77-LH-28-1 • AC-42 • AC-260,584 • Aceclidine • Acetylcholine • AF30 • AF150(S) • AF267B • AFDX-384 • Alvameline • AQRA-741 • Arecoline • Bethanechol • Butyrylcholine • Carbachol • CDD-0034 • CDD-0078 • CDD-0097 • CDD-0098 • CDD-0102 • Cevimeline • cis-Dioxolane • Ethoxysebacylcholine • LY-593,039 • L-689,660 • LY-2,033,298 • McNA343 • Methacholine • Milameline • Muscarine • NGX-267 • Ocvimeline • Oxotremorine • PD-151,832 • Pilocarpine • RS86 • Sabcomeline • SDZ 210-086 • Sebacylcholine • Suberylcholine • Talsaclidine • Tazomeline • Thiopilocarpine • Vedaclidine • VU-0029767 • VU-0090157 • VU-0152099 • VU-0152100 • VU-0238429 • WAY-132,983 • Xanomeline • YM-796
Antagonists: 3-Quinuclidinyl Benzilate • 4-DAMP • Aclidinium Bromide • Anisodamine • Anisodine • Atropine • Atropine Methonitrate • Benactyzine • Benzatropine (Benztropine) • Benzydamine • BIBN 99 • Biperiden • Bornaprine • CAR-226,086 • CAR-301,060 • CAR-302,196 • CAR-302,282 • CAR-302,368 • CAR-302,537 • CAR-302,668 • CS-27349 • Cyclobenzaprine • Cyclopentolate • Darifenacin • DAU-5884 • Dimethindene • Dexetimide • DIBD • Dicyclomine (Dicycloverine) • Ditran • EA-3167 • EA-3443 • EA-3580 • EA-3834 • Elemicin • Etanautine • Etybenzatropine (Ethylbenztropine) • Flavoxate • Himbacine • HL-031,120 • Ipratropium bromide • J-104,129 • Hyoscyamine • Mamba Toxin 3 • Mamba Toxin 7 • Mazaticol • Mebeverine • Methoctramine • Metixene • Myristicin • N-Ethyl-3-Piperidyl Benzilate • N-Methyl-3-Piperidyl Benzilate • Orphenadrine • Otenzepad • Oxybutynin • PBID • PD-102,807 • PD-0298029 • Phenglutarimide • Phenyltoloxamine • Pirenzepine • Piroheptine • Procyclidine • Profenamine • RU-47,213 • SCH-57,790 • SCH-72,788 • SCH-217,443 • Scopolamine (Hyoscine) • Solifenacin • Telenzepine • Tiotropium bromide • Tolterodine • Trihexyphenidyl • Tripitamine • Tropatepine • Tropicamide • WIN-2299 • Xanomeline • Zamifenacin; Others: 1st Generation Antihistamines (Brompheniramine, chlorphenamine, cyproheptadine, dimenhydrinate, diphenhydramine, doxylamine, mepyramine/pyrilamine, phenindamine, pheniramine, tripelennamine, triprolidine, etc) • Tricyclic Antidepressants (Amitriptyline, doxepin, trimipramine, etc) • Tetracyclic Antidepressants (Amoxapine, maprotiline, etc) • Typical Antipsychotics (Chlorpromazine, thioridazine, etc) • Atypical Antipsychotics (Clozapine, olanzapine, quetiapine, etc)Agonists: 5-HIAA • A-84,543 • A-366,833 • A-582,941 • A-867,744 • ABT-202 • ABT-418 • ABT-560 • ABT-894 • Acetylcholine • Altinicline • Anabasine • Anatoxin-a • AR-R17779 • Butyrylcholine • Carbachol • Cotinine • Cytisine • Decamethonium • Desformylflustrabromine • Dianicline • Dimethylphenylpiperazinium • Epibatidine • Epiboxidine • Ethanol • Ethoxysebacylcholine • EVP-4473 • EVP-6124 • Galantamine • GTS-21 • Ispronicline • Lobeline • MEM-63,908 (RG-3487) • Nicotine • NS-1738 • PHA-543,613 • PHA-709,829 • PNU-120,596 • PNU-282,987 • Pozanicline • Rivanicline • Sazetidine A • Sebacylcholine • SIB-1508Y • SIB-1553A • SSR-180,711 • Suberylcholine • TC-1698 • TC-1734 • TC-1827 • TC-2216 • TC-5214 • TC-5619 • TC-6683 • Tebanicline • Tropisetron • UB-165 • Varenicline • WAY-317,538 • XY-4083
Antagonists: 18-Methoxycoronaridine • α-Bungarotoxin • α-Conotoxin • Alcuronium • Amantadine • Anatruxonium • Atracurium • Bupropion (Amfebutamone) • Chandonium • Chlorisondamine • Cisatracurium • Coclaurine • Coronaridine • Dacuronium • Decamethonium • Dextromethorphan • Dextropropoxyphene • Dextrorphan • Diadonium • DHβE • Dimethyltubocurarine (Metocurine) • Dipyrandium • Dizocilpine (MK-801) • Doxacurium • Duador • Esketamine • Fazadinium • Gallamine • Hexafluronium • Hexamethonium (Benzohexonium) • Ibogaine • Isoflurane • Ketamine • Kynurenic acid • Laudexium (Laudolissin) • Levacetylmethadol • Malouetine • Mecamylamine • Memantine • Methadone • Methorphan (Racemethorphan) • Methyllycaconitine • Metocurine • Mivacurium • Morphanol (Racemorphanol) • Neramexane • Nitrous Oxide • Pancuronium • Pempidine • Pentamine • Pentolinium • Phencyclidine • Pipecuronium • Radafaxine • Rapacuronium • Rocuronium • Surugatoxin • Suxamethonium (Succinylcholine) • Thiocolchicoside • Toxiferine • Trimethaphan • Tropeinium • Tubocurarine • Vecuronium • XenonReuptake inhibitors PlasmalemmalCHT InhibitorsHemicholinium-3 (Hemicholine; HC3) • TriethylcholineVAChT InhibitorsEnzyme inhibitors ChAT inhibitors1-(-Benzoylethyl)pyridinium • 2-(α-Naphthoyl)ethyltrimethylammonium • 3-Chloro-4-stillbazole • 4-(1-Naphthylvinyl)pyridine • Acetylseco hemicholinium-3 • Acryloylcholine • AF64A • B115 • BETA • CM-54,903 • CatabolismAChE inhibitorsReversible: Carbamates: Aldicarb • Bendiocarb • Bufencarb • Carbaryl • Carbendazim • Carbetamide • Carbofuran • Chlorbufam • Chloropropham • Ethienocarb • Ethiofencarb • Fenobucarb • Fenoxycarb • Formetanate • Furadan • Ladostigil • Methiocarb • Methomyl • Miotine • Oxamyl • Phenmedipham • Pinmicarb • Pirimicarb • Propamocarb • Propham • Propoxur; Stigmines: Ganstigmine • Neostigmine • Phenserine • Physostigmine • Pyridostigmine • Rivastigmine; Others: Acotiamide • Ambenonium • Donepezil • Edrophonium • Galantamine • Huperzine A • Minaprine • Tacrine • Zanapezil
Irreversible: Organophosphates: Acephate • Azinphos-methyl • Bensulide • Cadusafos • Chlorethoxyfos • Chlorfenvinphos • Chlorpyrifos • Chlorpyrifos-Methyl • Coumaphos • Cyclosarin (GF) • Demeton • Demeton-S-Methyl • Diazinon • Dichlorvos • Dicrotophos • Diisopropyl fluorophosphate (Guthion) • Diisopropylphosphate • Dimethoate • Dioxathion • Disulfoton • EA-3148 • Echothiophate • Ethion • Ethoprop • Fenamiphos • Fenitrothion • Fenthion • Fosthiazate • GV • Isofluorophate • Isoxathion • Malaoxon • Malathion • Methamidophos • Methidathion • Metrifonate • Mevinphos • Monocrotophos • Naled • Novichok agent • Omethoate • Oxydemeton-Methyl • Paraoxon • Parathion • Parathion-Methyl • Phorate • Phosalone • Phosmet • Phostebupirim • Phoxim • Pirimiphos-Methyl • Sarin (GB) • Soman (GD) • Tabun (GA) • Temefos • Terbufos • Tetrachlorvinphos • Tribufos • Trichlorfon • VE • VG • VM • VR • VX; Others: Demecarium • Onchidal (Onchidella binneyi)BChE inhibitorsCymserine * Many of the acetylcholinesterase inhibitors listed above act as butyrylcholinesterase inhibitors.Others Choline (Lecithin) • Citicoline • Cyprodenate • Dimethylethanolamine (DMAE, deanol) • Glycerophosphocholine • Meclofenoxate (Centrophenoxine) • Phosphatidylcholine • Phosphatidylethanolamine • Phosphorylcholine • PirisudanolOthersAcetylcholine releasing agents: α-Latrotoxin • β-Bungarotoxin; Acetylcholine release inhibitors: Botulinum toxin (Botox); Acetylcholinesterase reactivators: Asoxime • Obidoxime • PralidoximeCategories:- Muscle relaxants
Wikimedia Foundation. 2010.
Look at other dictionaries:
Neuromuscular junction — Electron micrograph showing a cross section through the neuromuscular junction. T is the axon terminal, M is the muscle fiber. The arrow shows junctional folds … Wikipedia
drug — drug1 /drug/, n., v., drugged, drugging. n. 1. Pharm. a chemical substance used in the treatment, cure, prevention, or diagnosis of disease or used to otherwise enhance physical or mental well being. 2. (in federal law) a. any substance… … Universalium
Drug — /droog/, n. Zoroastrianism. the cosmic principle of disorder and falsehood. Cf. Asha. [ < Avestan drauga] * * * I Any chemical agent that affects the function of living things. Some, including antibiotics, stimulants, tranquilizers,… … Universalium
blocking agent — noun a class of drugs that inhibit (block) some biological process • Syn: ↑blocker • Derivationally related forms: ↑block (for: ↑blocker) • Hypernyms: ↑medicine, ↑ … Useful english dictionary
Mivacurium chloride — Systematic (IUPAC) name (1R,1 R) 2,2 [[(4E) 1,8 dioxooct 4 ene 1,8 diyl]bis(oxypropane 3,1 diyl)]bis[6,7 dimethoxy 2 methyl 1 (3,4,5 trimethoxybenzyl) 1,2,3,4 tetrahydroisoquinolinium] … Wikipedia
Candocuronium iodide — Systematic (IUPAC) name (4aS,4bR,8S,10aR,10bS,12aS) 1,1,10a,12a tetramethyl 8 (1 methylpyrrolidin 1 ium 1 yl) 3,4,4a,4b,5,7,8,9,10,10b,11,12 dodecahydro 2H naphtho[2,1 f]quinolin 1 ium diiodide … Wikipedia
Tubocurarine chloride — Systematic (IUPAC) name 6,6 dimethoxy 2,2 … Wikipedia
Doxacurium chloride — Systematic (IUPAC) name bis[3 [6,7,8 trimethoxy 2 methyl 1 [(3,4,5 trimethoxyphenyl)methyl] 3,4 dihydro 1H isoquinolin 2 yl] propyl] butanedioate dichloride Clinical da … Wikipedia
Sugammadex — Clinical data … Wikipedia
Cisatracurium besilate — Systematic (IUPAC) name 5 [3 [(1R,2R) 1 [(3,4 dimethoxyphenyl)methyl] 6,7 dimethoxy 2 methyl 3,4 dihydro 1H isoquinolin 2 yl] propanoyloxy]pentyl 3 [(1R,2R) 1 [(3,4 dimethoxy phenyl)methyl] 6,7 dimethoxy 2 methyl 3,4 … Wikipedia
 18+© Academic, 2000-2024
18+© Academic, 2000-2024- Contact us: Technical Support, Advertising
Dictionaries export, created on PHP, Joomla, Drupal, WordPress, MODx.