- Cisatracurium besilate
-
Cisatracurium besilate 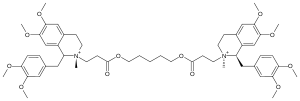
Systematic (IUPAC) name 5-[3-[(1R,2R)-1-[(3,4-dimethoxyphenyl)methyl]-6,7-
dimethoxy-2-methyl-3,4-dihydro-1H-isoquinolin-2-yl]
propanoyloxy]pentyl 3-[(1R,2R)-1-[(3,4-dimethoxy
phenyl)methyl]-6,7-dimethoxy-2-methyl-3,4-dihydro-
1H-isoquinolin-2-yl]propanoateClinical data AHFS/Drugs.com monograph Pregnancy cat. B Legal status Worldwide: prescription-only medicine Routes IV use only Pharmacokinetic data Bioavailability 100% (IV) Metabolism 80% Hoffman degradation/ Hepatic Half-life 20–29 minutes Excretion 10-15% unchanged Identifiers CAS number 96946-42-8 
ATC code M03AC11 PubChem CID 62886 DrugBank APRD00874 ChemSpider 56614 UNII 80YS8O1MBS ChEBI CHEBI:3721 ChEMBL CHEMBL1201248 
Chemical data Formula C53H72N2O12+2 Mol. mass 929.145 g/mol SMILES eMolecules & PubChem - InChI=1S/C53H72N2O12.2C6H6O3S/c1-54(22-18-38-32-48(62-7)50(64-9)34-40(38)42(54)28-36-14-16-44(58-3)46(30-36)60-5)24-20-52(56)66-26-12-11-13-27-67-53(57)21-25-55(2)23-19-39-33-49(63-8)51(65-10)35-41(39)43(55)29-37-15-17-45(59-4)47(31-37)61-6;2*7-10(8,9)6-4-2-1-3-5-6/h14-17,30-35,42-43H,11-13,18-29H2,1-10H3;2*1-5H,(H,7,8,9)/q+2;;/p-2/t42-,43-,54-,55-;;/m1../s1
Key:XXZSQOVSEBAPGS-DONVQRBFSA-L
 (what is this?) besilate (verify)
(what is this?) besilate (verify)Cisatracurium (formerly recognized as 51W89,[1] and marketed as Nimbex) is a neuromuscular-blocking drug or skeletal muscle relaxant in the category of non-depolarizing neuromuscular-blocking drugs, used adjunctively in anesthesia to facilitate endotracheal intubation and to provide skeletal muscle relaxation during surgery or mechanical ventilation. It is a bisbenzyltetrahydroisoquinolinium agent with an intermediate duration of action. Cisatracurium is one of the ten isomers of the parent molecule, atracurium.[2] Moreover, cisatracurium represents approximately 15% of the atracurium mixture.[3]
Contents
History
The generic name cisatracurium was conceived by scientists at Burroughs Wellcome Co. (now part of GlaxoSmithKline) by combining the name "atracurium" with "cis" [hence cisatracurium] because the molecule is the cis-cis isomer comprising the ten isomers of the parent, atracurium.[2] Atracurium itself was invented at Strathclyde University and licensed to Burroughs Wellcome Co., Research Triangle Park, NC, for further development and subsequent marketing as Tracrium. As the secondary pharmacology of atracurium was being developed, it became clear that the primary clinical disadvantage of atracurium was likely to be its propensity to elicit histamine release. To address this issue, a program was initiated to investigate the individual isomer constituents of atracurium to identify and isolate the isomer(s) associated with the undesirable histamine effects as well as identify the isomer that might possibly retain the desirable properties without the histamine release. Thus, in 1989, D A Hill and G L Turner, PhD (both chemists at Wellcome, Dartford, UK) first synthesized cisatracurium as an individual molecule in its own right. The pharmacological research of these individual isomers[4] was then developed further primarily by R. Brandt Maehr and William B. Wastila, PhD (both of whom were pharmacologists within the Division of Pharmacology at Burroughs Wellcome Co.) in collaboration with John J. Savarese MD (who at the time was an anesthesiologist in the Dept. of Anesthesia, Harvard Medical School at the Massachusetts General Hospital, Boston, MA). Thereafter, the entire clinical development of cisatracurium was completed in a record short period from 1992 to 1994: the team of scientists was led by J. Neal Weakly PhD, Martha M. Abou-Donia PhD, and Steve Quessy PhD, in the Division of Clinical Neurosciences at Burroughs Wellcome Co., Research Triangle Park, NC. By the time of its approval for human use, in 1995, by the US Food and Drug Administration, Burroughs Wellcome Co. had merged with Glaxo Inc., and cisatracurium was approved to be marketed as Nimbex by GlaxoWellcome Inc. The trade name "Nimbex" was derived from inserting an "i" to the original proposal "Nmbex," which stood for excellent Neuromuscular blocker.
Owing to the intense marketing rivalry, during the 1980s and 1990s, between the two major competitors at the time in this area of anesthesia (i.e., Organon Inc. and Burroughs Wellcome, Co.), it was not surprising then that the knives of scrutiny and skepticism[5] were already being sharpened against cisatracurium in the court of peer-reviewed publications even before the voluminous documentation necessary for impartial review and approval of the drug had barely been dispatched to the desks at the US Food and Drug Administration.
Preclinical Pharmacology
In vitro studies using human plasma indicated that cisatracurium spontaneously degrades at physiological pH via Hofmann elimination to yield laudanosine and the quaternary monoacrylate. Subsequent ester hydrolysis of the monoacrylate generates the monoquaternary alcohol, although the rate-liminting step is Hofmann elimination.[3] In rat plasma, cisatracurium is also metabolized by non-specific carboxylesterases (a rate-limiting step) to the monoquaternary alcohol and the monoquaternary acid.[3]
Clinical pharmacology
As is evident with the parent molecule, atracurium,[6][7] cisatracurium is also susceptible to degradation by Hofmann elimination and ester hydrolysis as components of the in vivo metabolic processes.[citation needed] See the atracurium page for information on Hofmann elimination in vivo versus the Hofmann degradation chemical reaction.
Because Hofmann elimination is a temperature- and plasma pH-dependent process, cisatracurium's rate of degradation in vivo is highly influenced by body pH and temperature just as it is with the parent molecule, atracurium: thus, an increase in body pH favors the elimination process,[citation needed] whereas a decrease in temperature slows down the process.[citation needed]
One of the metabolites of cistracurium via Hofmann elimination is laudanosine - see the atracurium page for further discussion of the issue regarding this metabolite and its undesirable CNS effects. 80% of cisatracurium is metabolized eventually to laudanosine and 20% is metabolized hepatically or excreted renally.[citation needed] 10-15% of the dose is excreted unchanged in the urine.[citation needed]
Since Hofmann elimination is an organ-independent chemodegradative mechanism, there is little or no risk to the use of cisatracurium in patients with liver or renal disease when compared with other neuromuscular-blocking agents.[citation needed]
Adverse effects
Histamine release - hypotension, reflex tachycardia and cutaneous flush
Unlike the parent, atracurium, cisatracurium affords a much better pharmacological profile with respect to eliciting histamine release.
Bronchospasm - Pulmonary compliance
To date, cisatracurium has not been reported to elicit bronochospasm at doses that are clinically prescribed.
Laudanosine - Epileptic foci
Cisatracurium undergoes Hofmann elimination as a primary route of chemodegradation: consequently one of the metabolites from this process is laudanosine, a tertiary amino alkaloid reported to be a modest CNS stimulant with epileptogenic activity[8] and cardiovascular effects such a hypotension and bradycardia.[9]. As a tertiary amine, Laudanosine is unionised and readily crosses the blood-brain barrier. Presently, there is little evidence that laudanosine accumulation and related toxicity will likely ever be seen with the doses of cisatracurium that are administered in clinical practice especially given that the plasma concentrations of laudanosine generated are lower with cisatracurium than those seen with atracurium.[9]
References
- ^ Meretoja OA, Taivainen T, Wirtavuori K (Jan 1995). "Pharmacodynamic effects of 51W89, an isomer of atracurium, in children during halothane anaesthesia". Br J Anaesth 74 (1): 6–11. doi:10.1093/bja/74.1.6. PMID 7880708.
- ^ a b Stenlake JB, Waigh RD, Dewar GH, Dhar NC, Hughes R, Chapple DJ, Lindon JC, Ferrige AG. (1984). "Biodegradable neuromuscular blocking agents. Part 6. Stereochemical studies on atracurium and related polyalkylene di-esters.". Eur J Med Chem 19 (5): 441–450.
- ^ a b c Dear GJ, Harrelson JC, Jones AE, Johnson TE, Pleasance S (1995). "Identification of urinary and biliary conjugated metabolites of the neuromuscular blocker 51W89 by liquid chromatography/mass spectrometry". Rapid Commun Mass Spectrom 9 (14): 1457–1464. doi:10.1002/rcm.1290091425. PMID 8534894.
- ^ Wastila WB, Maehr RB, Turner GL, Hill DA, Savarese JJ (Jul 1996). "Comparative pharmacology of cisatracurium (51W89), atracurium, and five isomers in cats". Anesthesiol 85 (1): 169–177. doi:10.1097/00000542-199607000-00023. PMID 8694363.
- ^ Miller RD (Jan 1995). "Is 51W89 an improvement compared with atracurium?". Br J Anaesth 74 (1): 1–2. doi:10.1093/bja/74.1.1. PMID 7533512.
- ^ Stiller RL, Cook DR, Chakravorti S. (1985). "In vitro degradation of atracurium in human plasma". Br J Anaesth 57 (11): 1085–1088. doi:10.1093/bja/57.11.1085. PMID 3840382.
- ^ Nigrovic V, Fox JL. (1991). "Atracurium decay and the formation of laudanosine in humans". Anesthesiol 74 (3): 446–454. doi:10.1097/00000542-199103000-00010. PMID 2001023.
- ^ Standaert FG (Dec 1985). "Magic bullets, science, and medicine". Anesthesiol 63 (6): 577–578. doi:10.1097/00000542-198512000-00002. PMID 2932980.
- ^ a b Fodale V, Santamaria LB (Jul 2002). "Laudanosine, an atracurium and cisatracurium metabolite". Eur J Anaesthesiol 19 (7): 466–473. PMID 12113608.
Further reading
- Caldwell JE (1995). "New skeletal muscle relaxants". Int Anesthesiol Clin 33 (1): 39–60. doi:10.1097/00004311-199500000-00003. PMID 7635557.
- Hull CJ (1995). "Pharmacokinetics and pharmacodynamics of the benzylisoquinolinium muscle relaxants". Acta Anaesthesiol Scand 106 Suppl: 13–17. PMID 8533537.
- Savarese JJ, Wastila WB (1995). "The future of the benzylisoquinolinium relaxants". Acta Anaesthesiol Scand 106 Suppl: 91–93. PMID 8533554.
- Esmaoglu A, Akin A, Mizrak A, Turk Y, Boyaci A (2006). "Addition of cisatracurium to lidocaine for intravenous regional anesthesia.". J Clin Anesth 18 (3): 194–7. doi:10.1016/j.jclinane.2005.08.003. PMID 16731321.
- Melloni C, De Vivo P, Launo C, Mastronardi P, Novelli G, Romano E (2006). "Cisatracurium versus vecuronium: a comparative, double blind, randomized, multicenter study in adult patients under propofol/fentanyl/N2O anesthesia.". Minerva Anestesiol 72 (5): 299–308. PMID 16675938.
- Serra C, Oliveira A (2006). "Cisatracurium: myographical and electrophysiological studies in the isolated rat muscle.". Fundam Clin Pharmacol 20 (3): 291–8. doi:10.1111/j.1472-8206.2006.00395.x. PMID 16671964.
Skeletal muscle relaxants (M03) Peripherally acting
(primarily antinicotinic,
NMJ block)Curare alkaloidsultra-short duration: Gantacurium
short duration: Mivacurium • Chandonium
intermediate duration: Atracurium • Cisatracurium • Fazadinium • Rocuronium • Vecuronium
long duration: Doxacurium • Dimethyltubocurarine • Pancuronium • Pipecuronium • Laudexium • Gallamine
unsorted: Hexafluronium (Hexafluorenium)Centrally acting Carbamic acid estersBenzodiazepinesAnticholinergics (Antimuscarinics)OtherBaclofen • Chlormezanone • Chlorphenesin • Chlorzoxazone • Donepezil • Eperisone • Flopropione • Mephenesin • Mephenoxalone • Metaxalone • Phenyramidol • Pridinol • Promoxolane • Quinine • Thiocolchicoside • Tizanidine • Tolperisone • TrazodoneDirectly acting Cholinergics Receptor ligands Agonists: 77-LH-28-1 • AC-42 • AC-260,584 • Aceclidine • Acetylcholine • AF30 • AF150(S) • AF267B • AFDX-384 • Alvameline • AQRA-741 • Arecoline • Bethanechol • Butyrylcholine • Carbachol • CDD-0034 • CDD-0078 • CDD-0097 • CDD-0098 • CDD-0102 • Cevimeline • cis-Dioxolane • Ethoxysebacylcholine • LY-593,039 • L-689,660 • LY-2,033,298 • McNA343 • Methacholine • Milameline • Muscarine • NGX-267 • Ocvimeline • Oxotremorine • PD-151,832 • Pilocarpine • RS86 • Sabcomeline • SDZ 210-086 • Sebacylcholine • Suberylcholine • Talsaclidine • Tazomeline • Thiopilocarpine • Vedaclidine • VU-0029767 • VU-0090157 • VU-0152099 • VU-0152100 • VU-0238429 • WAY-132,983 • Xanomeline • YM-796
Antagonists: 3-Quinuclidinyl Benzilate • 4-DAMP • Aclidinium Bromide • Anisodamine • Anisodine • Atropine • Atropine Methonitrate • Benactyzine • Benzatropine (Benztropine) • Benzydamine • BIBN 99 • Biperiden • Bornaprine • CAR-226,086 • CAR-301,060 • CAR-302,196 • CAR-302,282 • CAR-302,368 • CAR-302,537 • CAR-302,668 • CS-27349 • Cyclobenzaprine • Cyclopentolate • Darifenacin • DAU-5884 • Dimethindene • Dexetimide • DIBD • Dicyclomine (Dicycloverine) • Ditran • EA-3167 • EA-3443 • EA-3580 • EA-3834 • Elemicin • Etanautine • Etybenzatropine (Ethylbenztropine) • Flavoxate • Himbacine • HL-031,120 • Ipratropium bromide • J-104,129 • Hyoscyamine • Mamba Toxin 3 • Mamba Toxin 7 • Mazaticol • Mebeverine • Methoctramine • Metixene • Myristicin • N-Ethyl-3-Piperidyl Benzilate • N-Methyl-3-Piperidyl Benzilate • Orphenadrine • Otenzepad • Oxybutynin • PBID • PD-102,807 • PD-0298029 • Phenglutarimide • Phenyltoloxamine • Pirenzepine • Piroheptine • Procyclidine • Profenamine • RU-47,213 • SCH-57,790 • SCH-72,788 • SCH-217,443 • Scopolamine (Hyoscine) • Solifenacin • Telenzepine • Tiotropium bromide • Tolterodine • Trihexyphenidyl • Tripitamine • Tropatepine • Tropicamide • WIN-2299 • Xanomeline • Zamifenacin; Others: 1st Generation Antihistamines (Brompheniramine, chlorphenamine, cyproheptadine, dimenhydrinate, diphenhydramine, doxylamine, mepyramine/pyrilamine, phenindamine, pheniramine, tripelennamine, triprolidine, etc) • Tricyclic Antidepressants (Amitriptyline, doxepin, trimipramine, etc) • Tetracyclic Antidepressants (Amoxapine, maprotiline, etc) • Typical Antipsychotics (Chlorpromazine, thioridazine, etc) • Atypical Antipsychotics (Clozapine, olanzapine, quetiapine, etc)Agonists: 5-HIAA • A-84,543 • A-366,833 • A-582,941 • A-867,744 • ABT-202 • ABT-418 • ABT-560 • ABT-894 • Acetylcholine • Altinicline • Anabasine • Anatoxin-a • AR-R17779 • Butyrylcholine • Carbachol • Cotinine • Cytisine • Decamethonium • Desformylflustrabromine • Dianicline • Dimethylphenylpiperazinium • Epibatidine • Epiboxidine • Ethanol • Ethoxysebacylcholine • EVP-4473 • EVP-6124 • Galantamine • GTS-21 • Ispronicline • Lobeline • MEM-63,908 (RG-3487) • Nicotine • NS-1738 • PHA-543,613 • PHA-709,829 • PNU-120,596 • PNU-282,987 • Pozanicline • Rivanicline • Sazetidine A • Sebacylcholine • SIB-1508Y • SIB-1553A • SSR-180,711 • Suberylcholine • TC-1698 • TC-1734 • TC-1827 • TC-2216 • TC-5214 • TC-5619 • TC-6683 • Tebanicline • Tropisetron • UB-165 • Varenicline • WAY-317,538 • XY-4083
Antagonists: 18-Methoxycoronaridine • α-Bungarotoxin • α-Conotoxin • Alcuronium • Amantadine • Anatruxonium • Atracurium • Bupropion (Amfebutamone) • Chandonium • Chlorisondamine • Cisatracurium • Coclaurine • Coronaridine • Dacuronium • Decamethonium • Dextromethorphan • Dextropropoxyphene • Dextrorphan • Diadonium • DHβE • Dimethyltubocurarine (Metocurine) • Dipyrandium • Dizocilpine (MK-801) • Doxacurium • Duador • Esketamine • Fazadinium • Gallamine • Hexafluronium • Hexamethonium (Benzohexonium) • Ibogaine • Isoflurane • Ketamine • Kynurenic acid • Laudexium (Laudolissin) • Levacetylmethadol • Malouetine • Mecamylamine • Memantine • Methadone • Methorphan (Racemethorphan) • Methyllycaconitine • Metocurine • Mivacurium • Morphanol (Racemorphanol) • Neramexane • Nitrous Oxide • Pancuronium • Pempidine • Pentamine • Pentolinium • Phencyclidine • Pipecuronium • Radafaxine • Rapacuronium • Rocuronium • Surugatoxin • Suxamethonium (Succinylcholine) • Thiocolchicoside • Toxiferine • Trimethaphan • Tropeinium • Tubocurarine • Vecuronium • XenonReuptake inhibitors PlasmalemmalCHT InhibitorsVAChT InhibitorsEnzyme inhibitors ChAT inhibitors1-(-Benzoylethyl)pyridinium • 2-(α-Naphthoyl)ethyltrimethylammonium • 3-Chloro-4-stillbazole • 4-(1-Naphthylvinyl)pyridine • Acetylseco hemicholinium-3 • Acryloylcholine • AF64A • B115 • BETA • CM-54,903 • CatabolismAChE inhibitorsReversible: Carbamates: Aldicarb • Bendiocarb • Bufencarb • Carbaryl • Carbendazim • Carbetamide • Carbofuran • Chlorbufam • Chloropropham • Ethienocarb • Ethiofencarb • Fenobucarb • Fenoxycarb • Formetanate • Furadan • Ladostigil • Methiocarb • Methomyl • Miotine • Oxamyl • Phenmedipham • Pinmicarb • Pirimicarb • Propamocarb • Propham • Propoxur; Stigmines: Ganstigmine • Neostigmine • Phenserine • Physostigmine • Pyridostigmine • Rivastigmine; Others: Acotiamide • Ambenonium • Donepezil • Edrophonium • Galantamine • Huperzine A • Minaprine • Tacrine • Zanapezil
Irreversible: Organophosphates: Acephate • Azinphos-methyl • Bensulide • Cadusafos • Chlorethoxyfos • Chlorfenvinphos • Chlorpyrifos • Chlorpyrifos-Methyl • Coumaphos • Cyclosarin (GF) • Demeton • Demeton-S-Methyl • Diazinon • Dichlorvos • Dicrotophos • Diisopropyl fluorophosphate (Guthion) • Diisopropylphosphate • Dimethoate • Dioxathion • Disulfoton • EA-3148 • Echothiophate • Ethion • Ethoprop • Fenamiphos • Fenitrothion • Fenthion • Fosthiazate • GV • Isofluorophate • Isoxathion • Malaoxon • Malathion • Methamidophos • Methidathion • Metrifonate • Mevinphos • Monocrotophos • Naled • Novichok agent • Omethoate • Oxydemeton-Methyl • Paraoxon • Parathion • Parathion-Methyl • Phorate • Phosalone • Phosmet • Phostebupirim • Phoxim • Pirimiphos-Methyl • Sarin (GB) • Soman (GD) • Tabun (GA) • Temefos • Terbufos • Tetrachlorvinphos • Tribufos • Trichlorfon • VE • VG • VM • VR • VX; Others: Demecarium • Onchidal (Onchidella binneyi)BChE inhibitorsCymserine * Many of the acetylcholinesterase inhibitors listed above act as butyrylcholinesterase inhibitors.Others Choline (Lecithin) • Citicoline • Cyprodenate • Dimethylethanolamine (DMAE, deanol) • Glycerophosphocholine • Meclofenoxate (Centrophenoxine) • Phosphatidylcholine • Phosphatidylethanolamine • Phosphorylcholine • PirisudanolOthersAcetylcholine releasing agents: α-Latrotoxin • β-Bungarotoxin; Acetylcholine release inhibitors: Botulinum toxin (Botox); Acetylcholinesterase reactivators: Asoxime • Obidoxime • PralidoximeCategories:- Muscle relaxants
- Quaternary ammonium compounds
- Tetrahydroisoquinolines
- Phenol ethers
Wikimedia Foundation. 2010.
Look at other dictionaries:
Cisatracurium — drugbox IUPAC name = 5 [3 [(1 R ,2 R ) 1 [(3,4 dimethoxyphenyl)methyl] 6,7 dimethoxy 2 methyl 3,4 dihydro 1 H isoquinolin 2 yl] propanoyloxy] pentyl 3 [(1 R ,2 R ) 1 [(3,4 dimethoxy phenyl)methyl] 6,7 dimethoxy 2 methyl 3,4 dihydro 1 H… … Wikipedia
Cisatracurium — Général Nom IUPAC 5 [3 [(1R,2R) 1 [(3,4 diméthoxyphényl)méthyl] 6,7 diméthoxy 2 méthyl 3,4 dihydro 1H isoquinoline 2 yl]propanoyloxy]pentyl 3 [(1R,2R) 1 [(3,4 di … Wikipédia en Français
Нимбекс — Действующее вещество ›› Цисатракурия бесилат* (Cisatracurium besilate*) Латинское название Nimbex АТХ: ›› M03AC11 Цисатракурий Фармакологическая группа: н Холинолитики (миорелаксанты) Нозологическая классификация (МКБ 10) ›› Z100* КЛАСС XXII… … Словарь медицинских препаратов
96946-42-8 — Cisatracurium Cisatracurium Général Nom IUPAC 5 [3 [(1R,2R) 1 [(3,4 diméthoxyphényl)méthyl] 6,7 diméthoxy 2 méthyl 3,4 dihydro 1H isoquinoline 2 yl]propanoyloxy]pentyl 3 [(1 … Wikipédia en Français
- InChI=1S/C53H72N2O12.2C6H6O3S/c1-54(22-18-38-32-48(62-7)50(64-9)34-40(38)42(54)28-36-14-16-44(58-3)46(30-36)60-5)24-20-52(56)66-26-12-11-13-27-67-53(57)21-25-55(2)23-19-39-33-49(63-8)51(65-10)35-41(39)43(55)29-37-15-17-45(59-4)47(31-37)61-6;2*7-10(8,9)6-4-2-1-3-5-6/h14-17,30-35,42-43H,11-13,18-29H2,1-10H3;2*1-5H,(H,7,8,9)/q+2;;/p-2/t42-,43-,54-,55-;;/m1../s1
