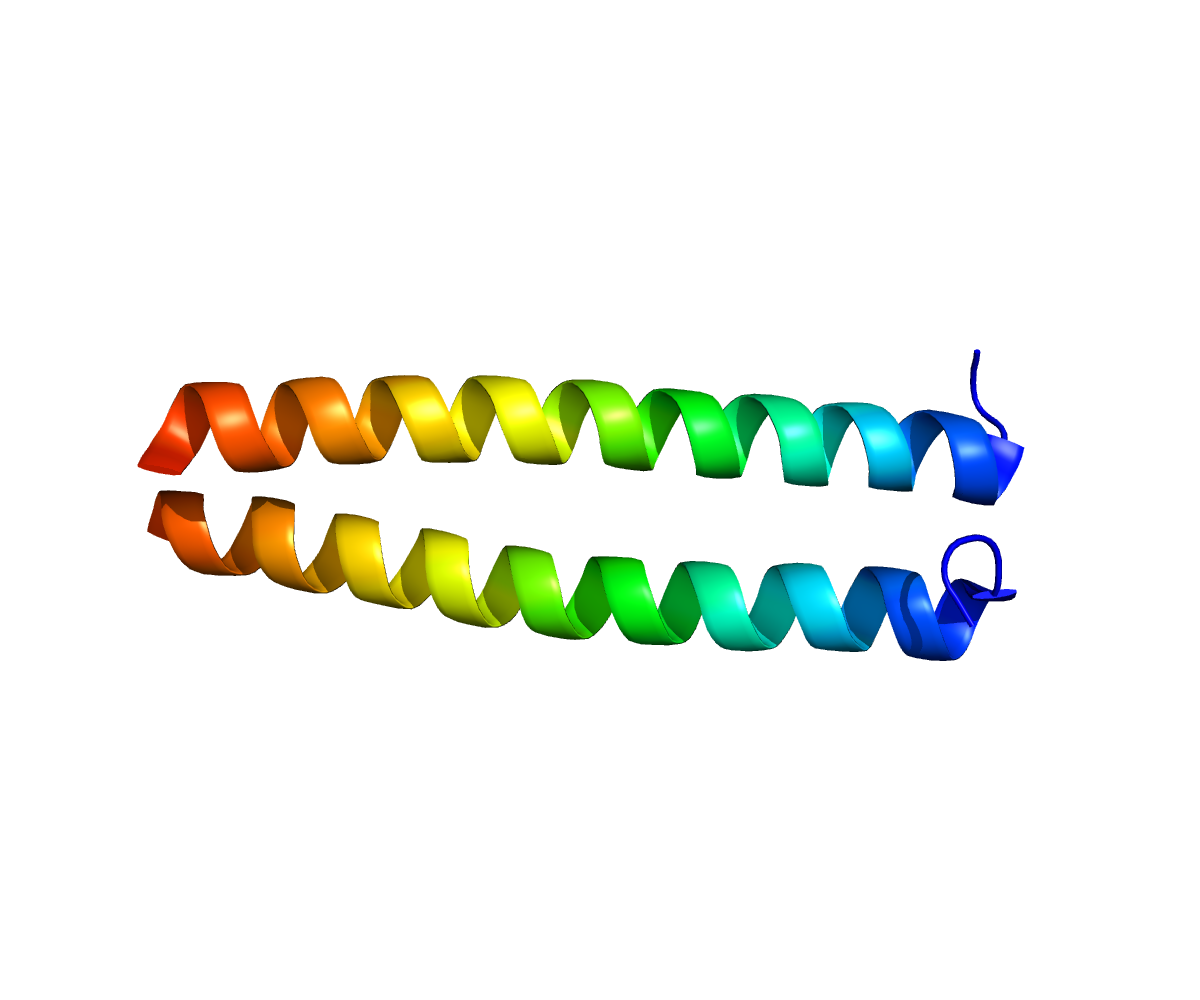- KvLQT1
-
Kv7.1 (KvLQT1) is a potassium channel protein coded for by the gene KCNQ1.[1] Kv7.1 is present in the cell membranes of cardiac muscle tissue and in inner ear neurons among other tissues. In the cardiac cells, Kv7.1 mediates the IKs (or slow delayed rectifying K+) current that contributes to the repolarization of the cell, terminating the cardiac action potential and thereby the heart's contraction.
Mutations in the gene can lead to a defective protein and several forms of inherited arrhythmias as Long QT syndrome, Short QT syndrome, and Familial Atrial Fibrillation. Currents arising from Kv7.1 in over-expression systems have never been recapitulated in native tissues - Kv7.1 is always found in native tissues with a modulatory subunit. In cardiac tissue, these subunits comprise KCNE1 and yotiao. Though physiologically irrelevant, homotetrameric Kv7.1 channels also display a unique form of C-type inactivation that reaches equilibrium quickly, allowing KvLQT1 currents to plateau. This is different from the inactivation seen in A-type currents, which causes rapid current decay.
This gene encodes a protein for a voltage-gated potassium channel required for the repolarization phase of the cardiac action potential. The gene product can form heteromultimers with two other potassium channel proteins, KCNE1 and KCNE3. Mutations in this gene are associated with hereditary long QT syndrome, Romano-Ward syndrome, Jervell and Lange-Nielsen syndrome and familial atrial fibrillation. The gene is located in a region of chromosome 11 that contains a large number of contiguous genes that are abnormally imprinted in cancer and the Beckwith-Wiedemann syndrome. Two alternative transcripts encoding distinct isoforms have been described.[2]
Contents
Interactions
KvLQT1 has been shown to interact with PRKACA,[3] PPP1CA[3] and AKAP9.[3]
See also
References
- ^ Jespersen T, Grunnet M, Olesen SP (2005). "The KCNQ1 potassium channel: from gene to physiological function". Physiology (Bethesda) 20 (6): 408–16. doi:10.1152/physiol.00031.2005. PMID 16287990.
- ^ "Entrez Gene: KCNQ1 potassium voltage-gated channel, KQT-like subfamily, member 1". http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=3784.
- ^ a b c Marx, Steven O; Kurokawa Junko, Reiken Steven, Motoike Howard, D'Armiento Jeanine, Marks Andrew R, Kass Robert S (Jan. 2002). "Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel". Science (United States) 295 (5554): 496–9. doi:10.1126/science.1066843. PMID 11799244.
Further reading
- Tranebjaerg L, Bathen J, Tyson J, Bitner-Glindzicz M (2000). "Jervell and Lange-Nielsen syndrome: a Norwegian perspective". Am. J. Med. Genet. 89 (3): 137–46. doi:10.1002/(SICI)1096-8628(19990924)89:3<137::AID-AJMG4>3.0.CO;2-C. PMID 10704188.
- Gutman GA, Chandy KG, Grissmer S, et al. (2006). "International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels". Pharmacol. Rev. 57 (4): 473–508. doi:10.1124/pr.57.4.10. PMID 16382104.
External links
Ca2+: Calcium channel Ligand-gatedNa+: Sodium channel Constitutively activeProton gatedK+: Potassium channel Kvα1-6 (1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8) · (2.1, 2.2) · (3.1, 3.2, 3.3, 3.4) · (4.1, 4.2, 4.3) · (5.1) · (6.1, 6.2, 6.3, 6.4)
Kvα7-12 (7.1, 7.2, 7.3, 7.4, 7.5) · (8.1, 8.2) · (9.1, 9.2, 9.3) · (10.1, 10.2) · (11.1/hERG, 11.2, 11.3) · (12.1, 12.2, 12.3)
Kvβ (1, 2, 3) · KCNIP (1, 2, 3, 4) · minK/ISK · minK/ISK-like · MiRP (1, 2, 3) · Shaker geneOther HVCN1General
This membrane protein-related article is a stub. You can help Wikipedia by expanding it.