- Wilson's disease
-
Wilson's disease Classification and external resources 
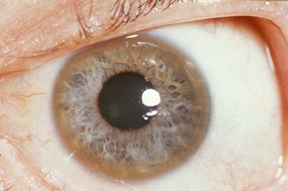
A Kayser-Fleischer ring (the brown ring on the edge of the iris) is common in Wilson's disease, especially when neurological symptoms are presentICD-10 E83.0 ICD-9 275.1 OMIM 277900 DiseasesDB 14152 MedlinePlus 000785 eMedicine med/2413 neuro/570 ped/2441 MeSH D006527 GeneReviews Wilson Disease Wilson's disease or hepatolenticular degeneration is an autosomal recessive genetic disorder in which copper accumulates in tissues; this manifests as neurological or psychiatric symptoms and liver disease. It is treated with medication that reduces copper absorption or removes the excess copper from the body, but occasionally a liver transplant is required.[1]
The condition is due to mutations in the Wilson disease protein (ATP7B) gene. A single abnormal copy of the gene is present in 1 in 100 people, who do not develop any symptoms (they are carriers). If a child inherits the gene from both parents, the child may develop Wilson's disease. Symptoms usually appear between the ages of 6 and 20 years, but cases in much older people have been described. Wilson's disease occurs in 1 to 4 per 100,000 people.[1] Wilson's disease is named after Samuel Alexander Kinnier Wilson (1878–1937), the British neurologist who first described the condition in 1912.[2]
Contents
Signs and symptoms
The main sites of copper accumulation are the liver and the brain, and consequently liver disease and neuropsychiatric symptoms are the main features that lead to diagnosis.[1] People with liver problems tend to come to medical attention earlier, generally as children or teenagers, than those with neurological and psychiatric symptoms, who tend to be in their twenties or older. Some are identified only because relatives have been diagnosed with Wilson's disease; many of these, when tested, turn out to have been experiencing symptoms of the condition but haven't received a diagnosis.[3]
Liver disease
Liver disease may present as tiredness, increased bleeding tendency or confusion (due to hepatic encephalopathy) and portal hypertension. The latter, a condition in which the pressure in the portal vein is markedly increased, leads to esophageal varices, blood vessels in the esophagus that may bleed in a life-threatening fashion, as well as enlargement of the spleen and accumulation of fluid in the abdominal cavity. On examination, signs of chronic liver disease such as spider naevi (small distended blood vessels, usually on the chest) may be observed. Chronic active hepatitis has caused cirrhosis of the liver in most by the time they develop symptoms. While most people with cirrhosis have an increased risk of hepatocellular carcinoma (liver cancer), this risk is relatively very low in Wilson's disease.[1]
About 5% of all people are diagnosed only when they develop fulminant acute liver failure, often in the context of a hemolytic anemia (anemia due to the destruction of red blood cells). This leads to abnormalities in protein production (identified by deranged coagulation) and metabolism by the liver. The deranged protein metabolism leads to the accumulation of waste products such as ammonia in the bloodstream. When these irritate the brain, the person develops hepatic encephalopathy (confusion, coma, seizures and finally life-threatening swelling of the brain).[1]
Neuropsychiatric symptoms
About half the people with Wilson's have neurological or psychiatric problems. Most initially have mild cognitive deterioration and clumsiness, as well as changes in behavior. Specific neurological symptoms then follow, often in the form of parkinsonism (cogwheel rigidity, bradykinesia or slowed movements and a lack of balance are the most common parkinsonism features[4]) with or without a typical hand tremor, masked facial expressions, slurred speech, ataxia (lack of coordination) or dystonia (twisting and repetitive movements of part of the body). Seizures and migraine appear to be more common in Wilson's disease.[1] Cognition can also be affected in Wilson's disease. This comes in two, not mutually exclusive, categories, frontal lobe disorder (may present as impulsivity, impaired judgement, promiscuity, apathy and executive dysfunction with poor planning and decision making) and subcortical dementia (may present as slow thinking, memory loss and executive dysfunction, without signs of aphasia, apraxia or agnosia). It is suggested that these cognitive involvements are related and closely linked to psychiatric manifestations of the disease.[4]
Psychiatric problems due to Wilson's disease may include behavioral changes, depression, anxiety and psychosis.[1] Psychiatric symptoms are commonly seen with the neurological symptoms, they rarely manifest on their own. These symptoms are often not well defined and can be attributed to other causes. Due to this, diagnosis of Wilson's disease is rarely made when only psychiatric symptoms are present.[4]
Other organ systems
Various medical conditions have been linked with copper accumulation in Wilson's disease:
- Eyes: Kayser–Fleischer rings (KF rings) may be visible around the iris. They are due to copper deposition in Descemet's membrane of the cornea. They do not occur in all people and may only be visible on slit lamp examination. Wilson's disease is also associated with sunflower cataracts, brown or green pigmentation of the anterior and posterior lens capsule. Neither cause significant visual loss.[1] KF rings occur in 66% of cases, more often in those with neurological than with liver problems.[3]
- Kidneys: renal tubular acidosis, a disorder of bicarbonate handling by the proximal tubules leads to nephrocalcinosis (calcium accumulation in the kidneys), weakening of the bone (due to calcium and phosphate loss) and occasionally aminoaciduria (loss of amino acids, needed for protein synthesis).[1]
- Heart: cardiomyopathy (weakness of the heart muscle) is a rare but recognized problem in Wilson's disease; it may lead to heart failure (fluid accumulation due to decreased pump function) and cardiac arrhythmias (episodes of irregular and/or abnormally fast or slow heart beat).[1]
- Hormones: hypoparathyroidism (failure of the parathyroid glands, leading to low calcium levels), infertility and habitual abortion.[1]
Genetics
 Wilson's disease has an autosomal recessive pattern of inheritance.
Wilson's disease has an autosomal recessive pattern of inheritance. Main article: ATP7B
Main article: ATP7BThe Wilson's disease gene (ATP7B) has been mapped to chromosome 13 (13q14.3) and is expressed primarily in the liver, kidney, and placenta. The gene codes for a P-type (cation transport enzyme) ATPase that transports copper into bile and incorporates it into ceruloplasmin.[1] Mutations can be detected in 90%. Most (60%) are homozygous for ATP7B mutations (two abnormal copies), and 30% have only one abnormal copy. Ten per cent have no detectable mutation.[3]
Although 300 mutations of ATP7B have been described, in most populations the cases of Wilson's disease are due to a small number of mutations specific for that population. For instance, in Western populations the H1069Q mutation (replacement of a histidine by a glutamine at position 1069 in the protein) is present in 37–63% of cases, while in China this mutation is very uncommon and R778L (arginine to leucine at 778) is found more often. Relatively little is known about the relative impact of various mutations, although the H1069Q mutation seems to predict later onset and predominantly neurological problems, according to some studies.[1][5]
A normal variation in the PRNP gene can modify the course of the disease by delaying the age of onset and affecting the type of symptoms that develop. This gene produces prion protein, which is active in the brain and other tissues and also appears to be involved in transporting copper.[6] A role for the ApoE gene was initially suspected but could not be confirmed.[5]
The condition is inherited in an autosomal recessive pattern. In order to inherit it, both of the parents of an individual must carry an affected gene. Most have no family history of the condition.[5] People with only one abnormal gene are called carriers (heterozygotes) and may have mild, but medically insignificant, abnormalities of copper metabolism.[7]
Wilson's disease is the most common of a group of hereditary diseases that cause copper overload in the liver. All can cause cirrhosis at a young age. The other members of the group are Indian childhood cirrhosis (ICC), endemic Tyrolean infantile cirrhosis and idiopathic copper toxicosis. These are not related to ATP7B mutations: for example, ICC has been linked to mutations in the KRT8 and the KRT18 gene.[5]
Pathophysiology
 Normal absorption and distribution of copper. Cu = copper, CP = ceruloplasmin, green = ATP7B carrying copper.
Normal absorption and distribution of copper. Cu = copper, CP = ceruloplasmin, green = ATP7B carrying copper.Copper is needed by the body for a number of functions, predominantly as a cofactor for a number of enzymes such as ceruloplasmin, cytochrome c oxidase, dopamine β-hydroxylase, superoxide dismutase and tyrosinase.[5]
Copper enters the body through the digestive tract. A transporter protein on the cells of the small bowel, copper membrane transporter 1 (CMT1), carries copper inside the cells, where some is bound to metallothionein and part is carried by ATOX1 to an organelle known as the trans-Golgi network. Here, in response to rising concentrations of copper, an enzyme called ATP7A releases copper into the portal vein to the liver. Liver cells also carry the CMT1 protein, and metallothionein and ATOX1 bind it inside the cell, but here it is ATP7B that links copper to ceruloplasmin and releases it into the bloodstream, as well as removing excess copper by secreting it into bile. Both functions of ATP7B are impaired in Wilson's disease. Copper accumulates in the liver tissue; ceruloplasmin is still secreted, but in a form that lacks copper (termed apoceruloplasmin) and rapidly degraded in the bloodstream.[5]
When the amount of copper in the liver overwhelms the proteins that normally bind it, it causes oxidative damage through a process known as Fenton chemistry; this damage eventually leads to chronic active hepatitis, fibrosis (deposition of connective tissue) and cirrhosis. The liver also releases copper into the bloodstream that is not bound to ceruloplasmin. This free copper precipitates throughout the body but particularly in the kidneys, eyes and brain. In the brain, most copper is deposited in the basal ganglia, particularly in the putamen and globus pallidus (together called the lenticular nucleus); these areas normally participate in the coordination of movement as well as playing a significant role in neurocognitive processes such as the processing of stimuli and mood regulation. Damage to these areas, again by Fenton chemistry, produces the neuropsychiatric symptoms seen in Wilson's disease.[5]
It is not clear why Wilson's disease causes hemolysis, but various lines of evidence suggest that high levels of free (non-ceruloplasmin bound) copper have a direct effect on either oxidation of hemoglobin, inhibition of energy-supplying enzymes in the red blood cell, or direct damage to the cell membrane.[8]
Diagnosis
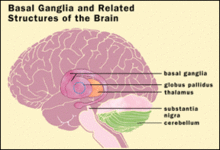 Location of the basal ganglia, the part of the brain affected by Wilson's disease
Location of the basal ganglia, the part of the brain affected by Wilson's diseaseWilson's disease may be suspected on the basis of any of the symptoms mentioned above, or when a close relative has been found to have Wilson's. Most have slightly abnormal liver function tests such as a raised aspartate transaminase, alanine transaminase and bilirubin level. If the liver damage is significant, albumin may be decreased due to an inability of damaged liver cells to produce this protein; likewise, the prothrombin time (a test of coagulation) may be prolonged as the liver is unable to produce proteins known as clotting factors.[1] Alkaline phosphatase levels are relatively low in those with Wilson's-related acute liver failure.[9] If there are neurological symptoms, magnetic resonance imaging (MRI) of the brain is usually performed; this shows hyperintensities in the part of the brain called the basal ganglia in the T2 setting.[7] MRI may also demonstrate the characteristic "face of the giant panda" pattern.[10]
There is no totally reliable test for Wilson's disease, but levels of ceruloplasmin and copper in the blood, as well of the amount of copper excreted in urine during a 24-hour period, are together used to form an impression of the amount of copper in the body. The gold standard or most ideal test is a liver biopsy.[1]
Ceruloplasmin
 Ceruloplasmin
CeruloplasminLevels of ceruloplasmin are abnormally low (<0.2 g/L) in 80–95% of cases.[1] It can, however, be present at normal levels in people with ongoing inflammation as it is an acute phase protein. Low ceruloplasmin is also found in Menkes disease and aceruloplasminemia, which are related to, but much rarer than Wilson's disease.[1][7]
The combination of neurological symptoms, Kayser–Fleisher rings and a low ceruloplasmin level is considered sufficient for the diagnosis of Wilson's disease. In many cases, however, further tests are needed.[7]
Serum and urine copper
Serum copper is paradoxically low but urine copper is elevated in Wilson's disease. Urine is collected for 24 hours in a bottle with a copper-free liner. Levels above 100 μg/24h (1.6 μmol/24h) confirm Wilson's disease, and levels above 40 μg/24h (0.6 μmol/24h) are strongly indicative.[1] High urine copper levels are not unique to Wilson's disease; they are sometimes observed in autoimmune hepatitis and in cholestasis (any disease obstructing the flow of bile from the liver to the small bowel).[7]
In children, the penicillamine test may be used. A 500 mg oral dose of penicillamine is administered, and urine collected for 24 hours. If this contains more than 1600 μg (25 μmol), it is a reliable indicator of Wilson's disease. This test has not been validated in adults.[7]
Liver biopsy
Once other investigations have indicated Wilson's disease, the ideal test is the removal of a small amount of liver tissue through a liver biopsy. This is assessed microscopically for the degree of steatosis and cirrhosis, and histochemistry and quantification of copper are used to measure the severity of the copper accumulation. A level of 250 μg of copper per gram of dried liver tissue confirms Wilson's disease. Occasionally, lower levels of copper are found; in that case, the combination of the biopsy findings with all other tests could still lead to a formal diagnosis of Wilson's.[1]
In the earlier stages of the disease, the biopsy typically shows steatosis (deposition of fatty material), increased glycogen in the nucleus, and areas of necrosis (cell death). In more advanced disease, the changes observed are quite similar to those seen in autoimmune hepatitis, such as infiltration by inflammatory cells, piecemeal necrosis and fibrosis (scar tissue). In advanced disease, finally, cirrhosis is the main finding. In acute liver failure, degeneration of the liver cells and collapse of the liver tissue architecture is seen, typically on a background of cirrhotic changes. Histochemical methods for detecting copper are inconsistent and unreliable, and taken alone are regarded as insufficient to establish a diagnosis.[7]
Genetic testing
Mutation analysis of the ATP7B gene, as well as other genes linked to copper accumulation in the liver, may be performed. Once a mutation is confirmed, it is possible to screen family members for the disease as part of clinical genetics family counselling.[1]
Treatment
Dietary
In general, a diet low in copper-containing foods is recommended, with the avoidance of mushrooms, nuts, chocolate, dried fruit, liver, and shellfish.[1]
Medication
Various treatments are available for Wilson's disease. Some increase the removal of copper from the body, while others prevent the absorption of copper from the diet.
Generally, penicillamine is the first treatment used. This binds copper (chelation) and leads to excretion of copper in the urine. Hence, monitoring of the amount of copper in the urine can be done to ensure a sufficiently high dose is taken. Penicillamine is not without problems: about 20% experience a side effect or complication of penicillamine treatment, such as drug-induced lupus (causing joint pains and a skin rash) or myasthenia (a nerve condition leading to muscle weakness). In those who presented with neurological symptoms, almost half experience a paradoxical worsening in their symptoms. While this phenomenon is also observed in other treatments for Wilson's, it is usually taken as an indication for discontinuing penicillamine and commencing second-line treatment.[1][7] Intolerant to penicillamine may instead be commenced on trientine hydrochloride, which also has chelating properties. Some recommend trientine as first-line treatment, but experience with penicillamine is more extensive.[7] A further agent with known activity in Wilson's disease is tetrathiomolybdate. This is still regarded as experimental,[7] although some studies have shown a beneficial effect.[1]
Once all results have returned to normal, zinc (usually in the form of a zinc acetate prescription called Galzin) may be used instead of chelators to maintain stable copper levels in the body. Zinc stimulates metallothionein, a protein in gut cells that binds copper and prevents their absorption and transport to the liver. Zinc therapy is continued unless symptoms recur, or if the urinary excretion of copper increases.[7]
In rare cases where none of the oral treatments are effective, especially in severe neurological disease, dimercaprol (British anti-Lewisite) is still occasionally necessary. This treatment is injected intramuscularly (into a muscle) every few weeks, and has a number of unpleasant side effects such as pain.[11]
People who are asymptomatic (for instance those diagnosed through family screening or only as a result of abnormal test results) are generally treated, as the copper accumulation may cause long-term damage in the future. It is unclear whether these people are best treated with penicillamine or zinc acetate.[7]
Physical therapy
Physiotherapy is beneficial for those patients with the neurologic form of the disease. The copper chelating treatment may take up to six months to start working, and physical therapy can assist in coping with ataxia, dystonia, and tremors, as well as preventing the development of contractures that can result from dystonia.[12]
Transplantation
Liver transplantation is an effective cure for Wilson's disease, but is used only in particular scenarios because of the numerous risks and complications associated with the procedure. It is used mainly in people with fulminant liver failure who fail to respond to medical treatment, or in those with advanced chronic liver disease. Liver transplantation is avoided in severe neuropsychiatric illness, in which its benefit has not been demonstrated.[1][7]
History
The disease bears the name of the British physician Samuel Alexander Kinnier Wilson (1878–1937), a neurologist who described the condition, including the pathological changes in the brain and liver, in 1912.[2] Wilson's work had been predated by, and drew on, reports from German neurologist Carl Westphal (in 1883), who termed it "pseudosclerosis"; by the British neurologist William Gowers (in 1888); and by Adolph Strümpell (in 1898), who noted hepatic cirrhosis.[13] Neuropathologist John Nathaniel Cumings made the link with copper accumulation in both the liver and the brain in 1948.[14] The occurrence of hemolysis was noted in 1967.[15]
Cumings, and simultaneously the New Zealand neurologist Derek Denny-Brown, working in the United States, first reported effective treatment with metal chelator British anti-Lewisite in 1951.[16][17] This treatment had to be injected but was one of the first therapies available in the field of neurology, a field that classically was able to observe and diagnose but had few treatments to offer.[13][18] The first effective oral chelation agent, penicillamine, was discovered in 1956 by British neurologist John Walshe.[19] In 1982, Walshe also introduced trientine,[20] and was the first to develop tetrathiomolybdate for clinical use.[21] Zinc acetate therapy initially made its appearance in the Netherlands, where physicians Schouwink and Hoogenraad used it in 1961 and in the 1970s, respectively, but it was further developed later by Brewer and colleagues at the University of Michigan.[11][22]
The genetic basis of Wilson's disease and linkage to ATP7B mutations was elucidated in the 1980s and 1990s by several research groups.[23][24]
In other animals
Hereditary copper accumulation has been described in Bedlington Terriers,[25] where it generally only affects the liver. It is due to mutations in the COMMD1 (or MURR1) gene.[26] In non-Wilsonian copper accumulation states (such as Indian childhood cirrhosis), no COMMD1 mutations could be detected to explain their genetic origin.[27]
References
- ^ a b c d e f g h i j k l m n o p q r s t u v w x Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML (2007). "Wilson's disease". Lancet 369 (9559): 397–408. doi:10.1016/S0140-6736(07)60196-2. PMID 17276780.
- ^ a b Kinnier Wilson SA (1912). "Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver" (PDF). Brain 34 (1): 295–507. doi:10.1093/brain/34.4.295. http://brain.oxfordjournals.org/cgi/reprint/34/4/295.
- ^ a b c Merle U, Schaefer M, Ferenci P, Stremmel W (2007). "Clinical presentation, diagnosis and long‐term outcome of Wilson's disease: a cohort study". Gut 56 (1): 115–20. doi:10.1136/gut.2005.087262. PMC 1856673. PMID 16709660. http://gut.bmj.com/cgi/content/full/56/1/115.
- ^ a b c Lorincz MT (2010). "Neurologic Wilson's disease". Annals of the New York Academy of Sciences 1184: 173–87. doi:10.1111/j.1749-6632.2009.05109.x. PMID 20146697. http://onlinelibrary.wiley.com/doi/10.1111/j.1749-6632.2009.05109.x/full.
- ^ a b c d e f g de Bie P, Muller P, Wijmenga C, Klomp LW (November 2007). "Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes". J. Med. Genet. 44 (11): 673–88. doi:10.1136/jmg.2007.052746. PMC 2752173. PMID 17717039. http://jmg.bmj.com/cgi/content/full/44/11/673.
- ^ Grubenbecher S, Stüve O, Hefter H, Korth C (2006). "Prion protein gene codon 129 modulates clinical course of neurological Wilson disease". Neuroreport 17 (5): 549–52. doi:10.1097/01.wnr.0000209006.48105.90. PMID 16543824.
- ^ a b c d e f g h i j k l m Roberts EA, Schilsky ML (2003). "A practice guideline on Wilson disease" (PDF). Hepatology 37 (6): 1475–92. doi:10.1053/jhep.2003.50252. PMID 12774027. http://www3.interscience.wiley.com/cgi-bin/fulltext/106595824/PDFSTART.
- ^ Lee, GR (1999). "Chapter 48: acquired hemolytic anaemias resulting from direct effects of infectious, chemical or physical agents". In Lee GR, Foerster J, Lukens J et al.. Wintrobe's clinical hematology. vol 1 (10th ed.). Williams & Wilkins. pp. 1298. ISBN 0-683-18242-0.
- ^ Shaver WA, Bhatt H, Combes B (1986). "Low serum alkaline phosphatase activity in Wilson's disease". Hepatology 6 (5): 859–63. doi:10.1002/hep.1840060509. PMID 3758940.
- ^ Das SK, Ray K (September 2006). "Wilson's disease: an update". Nat Clin Pract Neurol 2 (9): 482–93. doi:10.1038/ncpneuro0291. PMID 16932613. http://www.nature.com/nrneurol/journal/v2/n9/full/ncpneuro0291.html.
- ^ a b Walshe JM (July 1996). "Treatment of Wilson's disease: the historical background". QJM 89 (7): 553–5. PMID 8759497.
- ^ Brewer GJ, Askari FK (2005). "Wilson's disease: clinical management and therapy". Journal of Hepatology 42 (Suppl 1): 13–21. doi:10.1016/j.jhep.2004.11.013. PMID 15777568. http://www.jhep-elsevier.com/article/PIIS0168827804005318/fulltext.
- ^ a b Robertson WM (February 2000). "Wilson's disease". Arch. Neurol. 57 (2): 276–7. doi:10.1001/archneur.57.2.276. PMID 10681092. http://archneur.ama-assn.org/cgi/content/full/57/2/276.
- ^ Cumings JN (1948). "The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration" (PDF). Brain 71 (Dec): 410–5. doi:10.1093/brain/71.4.410. PMID 18124738. http://brain.oxfordjournals.org/cgi/reprint/71/4/410.
- ^ McIntyre N, Clink HM, Levi AJ, Cumings JN, Sherlock S (February 1967). "Hemolytic anemia in Wilson's disease". N. Engl. J. Med. 276 (8): 439–44. doi:10.1056/NEJM196702232760804. PMID 6018274.
- ^ Cumings JN (March 1951). "The effects of B.A.L. in hepatolenticular degeneration". Brain 74 (1): 10–22. doi:10.1093/brain/74.1.10. PMID 14830662.
- ^ Denny-Brown D, Porter H (December 1951). "The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson's disease)". N. Engl. J. Med. 245 (24): 917–25. doi:10.1056/NEJM195112132452401. PMID 14882450.
- ^ Vilensky JA, Robertson WM, Gilman S (September 2002). "Denny-Brown, Wilson's disease, and BAL (British antilewisite [2,3-dimercaptopropanol])". Neurology 59 (6): 914–6. PMID 12297577.
- ^ Walshe JM (January 1956). "Wilson's disease; new oral therapy". Lancet 267 (6906): 25–6. doi:10.1016/S0140-6736(56)91859-1. PMID 13279157.
- ^ Walshe JM (March 1982). "Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride". Lancet 1 (8273): 643–7. doi:10.1016/S0140-6736(82)92201-2. PMID 6121964.
- ^ Harper PL, Walshe JM (December 1986). "Reversible pancytopenia secondary to treatment with tetrathiomolybdate". Br. J. Haematol. 64 (4): 851–3. doi:10.1111/j.1365-2141.1986.tb02250.x. PMID 3801328.
- ^ Brewer GJ (January 2000). "Recognition, diagnosis, and management of Wilson's disease". Proc. Soc. Exp. Biol. Med. 223 (1): 39–46. doi:10.1046/j.1525-1373.2000.22305.x. PMID 10632959. http://www.ebmonline.org/cgi/content/full/223/1/39.
- ^ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993). "The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene". Nat. Genet. 5 (4): 327–37. doi:10.1038/ng1293-327. PMID 8298639.
- ^ Tanzi RE, Petrukhin K, Chernov I, et al. (1993). "The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene". Nat. Genet. 5 (4): 344–50. doi:10.1038/ng1293-344. PMID 8298641.
- ^ Sternlieb I, Twedt DC, Johnson GF, et al. (1977). "Inherited copper toxicity of the liver in Bedlington terriers". Proc. R. Soc. Med. 70 Suppl 3 (Suppl 3): 8–9. PMC 1543595. PMID 122681. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1543595.
- ^ van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C (2002). "Identification of a new copper metabolism gene by positional cloning in a purebred dog population". Hum. Mol. Genet. 11 (2): 165–73. doi:10.1093/hmg/11.2.165. PMID 11809725. http://hmg.oxfordjournals.org/cgi/content/full/11/2/165.
- ^ Müller T, van de Sluis B, Zhernakova A, et al. (2003). "The canine copper toxicosis gene MURR1 does not cause non-Wilsonian hepatic copper toxicosis". J. Hepatol. 38 (2): 164–8. doi:10.1016/S0168-8278(02)00356-2. PMID 12547404.
External links
- Wilson's disease association international
- Wilson's disease UK
- Children's Liver Disease Foundation (UK)
- Wilson Disease Mutation Database (HUGO)
- Wilson's disease at Who Named It?
- Wilson disease at NLM Genetics Home Reference
- Wilson Disease at NIH/UW GeneTests
Inborn error of metal metabolism (E83, 275) Transition metal high: Primary iron overload disorder: Hemochromatosis/HFE1 · Juvenile/HFE2 · HFE3 · African iron overload/HFE4 · Aceruloplasminemia · Atransferrinemia · Hemosiderosisdeficiency: Iron deficiencyCuhigh: Copper toxicity · Wilson's diseaseZnhigh: Zinc toxicitydeficiency: Acrodermatitis enteropathicaElectrolyte see Template:Water-electrolyte imbalance and acid-base imbalancehigh: Hyperphosphatemiahigh: Hypermagnesemiadeficiency: Hypomagnesemiahigh: Hypercalcaemia · Milk-alkali syndrome (Burnett's) · Calcinosis (Calciphylaxis, Calcinosis cutis) · Calcification (Metastatic calcification, Dystrophic calcification) · Familial hypocalciuric hypercalcemiaM: NUT
cof, enz, met
noco, nuvi, sysi/epon, met
drug(A8/11/12)
Genetic disorder, membrane: ATPase disorders ATP1 ATP2 ATP2A1 (Brody myopathy) · ATP2A2 (Darier's disease, Acrokeratosis verruciformis) · ATP2C1 (Hailey–Hailey disease)ATP7 ATP13 ATP13A2 (Kufor-Rakeb syndrome)Other Categories:- Neurological disorders
- Hepatology
- Diseases of liver
- Autosomal recessive disorders
- Rare diseases



Wikimedia Foundation. 2010.