- Desmoplastic small-round-cell tumor
-
Desmoplastic small-round-cell tumor Classification and external resources 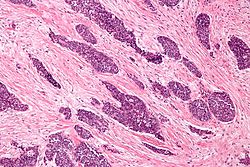
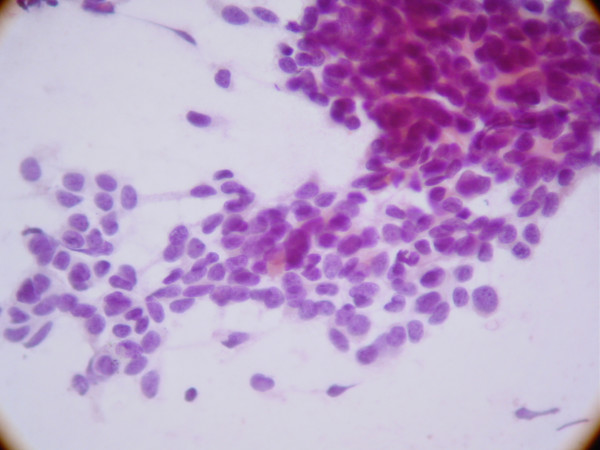
Micrograph of a desmoplastic small round cell tumor, showing the characteristic desmoplastic stroma and angulated nests of small round cells. H&E stain.ICD-O: 8806/3 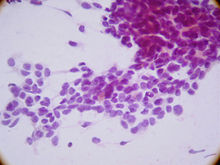 Display of small round blue cells characteristic of desmoplastic small-round-cell tumor.
Display of small round blue cells characteristic of desmoplastic small-round-cell tumor.
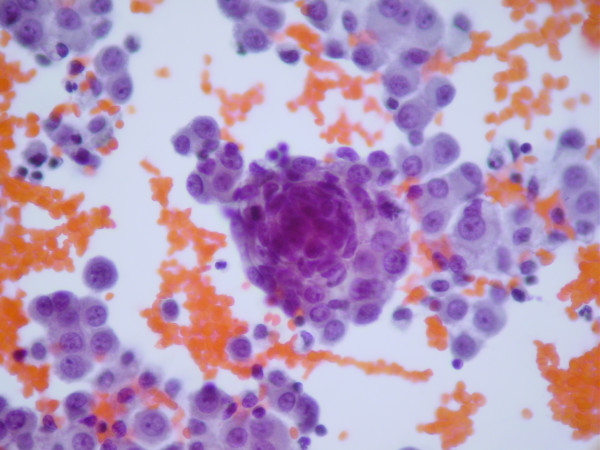
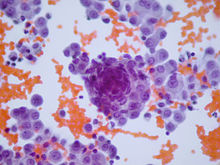 Cell exhibiting blue oval and round shapes of desmoplastic small-round blue cell tumor
Cell exhibiting blue oval and round shapes of desmoplastic small-round blue cell tumorDesmoplastic small-round-cell tumor is classified as a soft tissue sarcoma. It is an aggressive and rare tumor that primarily occurs as masses in the abdomen.[1] Other areas affected may include the lymph nodes, the lining of the abdomen, diaphragm, spleen, liver, chest wall, skull, spinal cord, large intestine, small intestine, bladder, brain, lungs, testicles, ovaries, and the pelvis. Reported sites of metatastic spread include the liver, lungs, lymph nodes, brain, skull, and bones.
The tumor is considered a childhood cancer that predominantly strikes boys and young adults.
The disease rarely occurs in females, but when it does the tumors can be mistaken for ovarian cancer.[2]
Contents
Causes
There are no known risk factors that have been identified specific to the disease. The tumor appears to arise from the primitive cells of childhood, and is considered a childhood cancer.
Research has indicated that there is a chimeric relationship between desmoplastic small-round-cell tumor (DSRCT) and Wilms' tumor and Ewing's sarcoma. Together with neuroblastoma and non-Hodgkin's lymphoma, they form the small or B cell tumors.
DSRCT is associated with a unique chromosomal translocation (t11;22)(p13:q12)[3] resulting in a EWS/WT1 transcript[4] that is diagnostic of this tumor.[1] This transcript codes for a protein that acts as a transcriptional activator that fails to suppress tumor growth.
The EWS/WT1 translocation product targets ENT4.[5] ENT4 is also known as PMAT.
Symptoms
There are few early warning signs that a patient has a DSRCT. Patients are often young and healthy as the tumors grow and spread uninhibited within the abdominal cavity. These are rare tumors and symptoms are often misdiagnosed by family physicians. The abdominal masses can grow to enormous size before being noticed by the patient. The tumors can be felt as hard, round masses by palpating the abdomen.
First symptoms of the disease often include abdominal distention, abdominal mass, abdominal or back pain, gastrointestinal obstruction, lack of appetite, ascites, anemia, and/or cachexia.
Other reported symptoms include unknown lumps, thyroid conditions, hormonal conditions, blood clotting, kidney or urological problems, testicle, breast, uterine, vaginal, or ovarian masses
Differential Diagnoses
Because this is a rare tumor, not many family physicians or oncologists are familiar with this disease. DSRCT in young patients can be mistaken for other abdominal tumors including rhabdomyosarcoma, neuroblastoma, and mesenteric carcinoid. In older patients DSRCT can resemble lymphoma, peritoneal mesothelioma, and peritoneal carcinomatosis. In males DSRCT may be mistaken for germ cell or testicular cancer while in females DSRCT can be mistaken for Ovarian cancer. DSRCT shares characteristics with other small-round blue cell cancers including Ewing's sarcoma, acute leukemia, small cell mesothelioma, neuroblastoma, primitive neuroectodermal tumor, rhabdomyosarcoma, and Wilms' tumor.
Pathology
The entity was first described by pathologists William L. Gerald and Juan Rosai in 1989.[6] Pathology reveals well circumscribed solid tumor nodules within a dense desmoplastic stroma. Often areas of central necrosis are present. Tumor cells have hyperchromatic nuclei with increased nuclear/cytoplasmic ratio.
On immunohistochemistry, these cells have trilinear coexpression including the epithelial marker cytokeratin, the mesenchymal markers desmin and vimentin, and the neuronal marker neuron-specific enolase. Thus, although initially thought to be of mesothelial origin due to sites of presentation, it is now hypothesized to arise from a progenitor cell with multiphenotypic differentiation.
Treatment
DSRCT is frequently misdiagnosed. Adult patients should always be referred to a sarcoma specialist. This is an aggressive, rare, fast spreading tumor and both pediatric and adult patients should be treated at a sarcoma center.
There is no standard protocol for the disease;[7] however, recent journals and studies have reported that some patients respond to high dose (P6 Protocol) chemotherapy, maintenance chemotherapy, debulking operation, cytoreductive surgery, and radiation therapy. Other treatment options include: hematopoietic stem cell transplantation, intensity-modulated radiation Therapy, radiofrequency ablation, stereotactic body radiation therapy, intraperitoneal hyperthermic chemoperfusion, and clinical trials.
Prognosis
The prognosis for DSRCT remains poor.[8] The 5-year survival rate of DRSCT is only approximately 15%.[9] Prognosis depends upon the stage of the cancer. Because the disease can be misdiagnosed or remain undetected, tumors frequently grow large within the abdomen and metastasized or seed to other parts of the body.
There is no known organ or area of origin. DSRCT can metastasize through lymph nodes or the blood stream. Sites of metastatis include the spleen, diaphragm, liver, large and small intestine, lungs, central nervous system, bones, uterus, bladder, genitals, abdominal cavity, and the brain.
A multi-modality approach of high dose chemotherapy, aggressive surgical resection,[7] radiation, and stem cell rescue improves survival for some patients. Reports have indicated that patients will initially respond to first line chemotherapy and treatment but that relapse is common.
Some patients in remission or with inoperable tumor seem to benefit from long term low dose chemotherapy, turning DSRCT into a chronic disease.
Research
The Stehlin Foundation[10] currently offers DSRCT patients the opportunity to send samples of their tumors free of charge for testing. Research scientists are growing the samples on nude mice and testing various chemical agents to find which are most effective against the individual's tumor.
Patients with advanced DSRCT may qualify to participate in clinical trials that are researching new drugs to treat the disease.
Alternative names
This disease is also known as: desmoplastic small round blue cell tumor; intraabdominal desmoplastic small round blue cell tumor; desmoplastic small cell tumor; desmoplastic cancer; desmoplastic sarcoma; DSRCT.
There is no connection to peritoneal mesothelioma which is another disease sometimes described as desmoplastic.
References
- ^ a b Lee YS, Hsiao CH (October 2007). "Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical and molecular study of four patients". J. Formos. Med. Assoc. 106 (10): 854–60. doi:10.1016/S0929-6646(08)60051-0. PMID 17964965. http://ajws.elsevier.com/ajws_pubmed/pubmed_switch.asp?journal_issn=0929-6646&art_pub_year=2007&art_pub_month=10&art_pub_vol=106&art_sp=854.
- ^ Bland AE, Shah AA, Piscitelli JT, Bentley RC, Secord AA (2007). "Desmoplastic small round cell tumor masquerading as advanced ovarian cancer". Int J Gynecol Cancer 18 (4): 847. doi:10.1111/j.1525-1438.2007.01110.x. PMID 18081791.
- ^ Murphy AJ, Bishop K, Pereira C, et al. (December 2008). "A new molecular variant of desmoplastic small round cell tumor: significance of WT1 immunostaining in this entity". Hum. Pathol. 39 (12): 1763–70. doi:10.1016/j.humpath.2008.04.019. PMID 18703217. http://linkinghub.elsevier.com/retrieve/pii/S0046-8177(08)00222-0.
- ^ Gerald WL, Haber DA (June 2005). "The EWS-WT1 gene fusion in desmoplastic small round cell tumor". Semin. Cancer Biol. 15 (3): 197–205. doi:10.1016/j.semcancer.2005.01.005. PMID 15826834. http://linkinghub.elsevier.com/retrieve/pii/S1044-579X(05)00006-4.
- ^ Li H, Smolen GA, Beers LF, et al. (2008). "Adenosine transporter ENT4 is a direct target of EWS/WT1 translocation product and is highly expressed in desmoplastic small round cell tumor". PLoS ONE 3 (6): e2353. doi:10.1371/journal.pone.0002353. PMC 2394657. PMID 18523561. http://www.plosone.org/article/info:doi/10.1371/journal.pone.0002353.
- ^ Gerald WL, Rosai J (1989). "Case 2. Desmoplastic small cell tumor with divergent differentiation". Pediatr Pathol 9 (2): 177–83. PMID 2473463.
- ^ a b Talarico F, Iusco D, Negri L, Belinelli D (2007). "Combined resection and multi-agent adjuvant chemotherapy for intra-abdominal desmoplastic small round cell tumour: case report and review of the literature". G Chir 28 (10): 367–70. PMID 17915050. http://www.giornalechirurgia.it/index.php?PAGE=article&ID=2393.
- ^ Aguilera D, Hayes-Jordan A, Anderson P, Woo S, Pearson M, Green H (2008). "Outpatient and home chemotherapy with novel local control strategies in DSRCT". Sarcoma 2008: 261589. doi:10.1155/2008/261589. PMC 2430011. PMID 18566684. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2430011.
- ^ Lal DR, Su WT, Wolden SL, Loh KC, Modak S, La Quaglia MP (January 2005). "Results of multimodal treatment for desmoplastic small round cell tumors". J. Pediatr. Surg. 40 (1): 251–5. doi:10.1016/j.jpedsurg.2004.09.046. PMID 15868593. http://linkinghub.elsevier.com/retrieve/pii/S0022346804006499.
- ^ Official website for Stehlin Foundation
External links
- humpath #2002 (Pathology images)
- Desmoplastic Small Round Cell Tumor in the Sarcoma Learning Center
- Desmoplastic Small Round Cell Tumor
- Desmoplastic Small Round Blue Cell Tumor
- Yahoo! Group for people afflicted with DSRCT, for patients to share treatment stories and tips with each other.
Connective/soft tissue tumors and sarcomas (ICD-O 8800–9059) (C45–C49/D17–D21, 171/214–215) Not otherwise specified (8800–8809) Connective tissue neoplasm Fibromatous (8810–8839)Fibroma/fibromatosis: Aggressive infantile fibromatosis · Aponeurotic fibroma · Collagenous fibroma · Diffuse infantile fibromatosis · Familial myxovascular fibromas · Fibroma of tendon sheath · Fibromatosis colli · Infantile digital fibromatosis · Juvenile hyaline fibromatosis · Plantar fibromatosis · Pleomorphic fibroma · Oral submucous fibrosisHistiocytoma/histiocytic sarcoma: Benign fibrous histiocytoma · Malignant fibrous histiocytoma · Atypical fibroxanthomaSolitary fibrous tumorMyxomatous (8840–8849)Myxoma/myxosarcoma (Cutaneous myxoma, Superficial acral fibromyxoma) · Angiomyxoma · Ossifying fibromyxoid tumourFibroepithelial (9000–9039)Synovial-like (9040–9049)Lipomatous (8850–8889) Chondroid lipoma · Intradermal spindle cell lipoma · Pleomorphic lipoma · Benign lipoblastomatosis · Spindle cell lipoma · HibernomaMyomatous (8890–8929) general: Myoma/myosarcomaskeletal muscle: Rhabdomyoma/rhabdomyosarcoma: Embryonal rhabdomyosarcoma (Sarcoma botryoides) · Alveolar rhabdomyosarcomaLeiomyoma · Angioleiomyoma · Angiolipoleiomyoma · Genital leiomyoma · Leiomyosarcoma · Multiple cutaneous and uterine leiomyomatosis syndrome · Multiple cutaneous leiomyoma · Neural fibrolipoma · Solitary cutaneous leiomyomaComplex mixed and stromal (8930–8999) Adenomyoma · Pleomorphic adenoma · Mixed Müllerian tumor · Mesoblastic nephroma · Wilms' tumor · Rhabdoid tumour · Clear-cell sarcoma of the kidney · Hepatoblastoma · Pancreatoblastoma · CarcinosarcomaMesothelial (9050–9059) see also Template:Connective tissueTumors: digestive system neoplasia (C15–C26/D12–D13, 150–159/211) GI tract Upper GI tractGastric carcinoma · Signet ring cell carcinoma · Gastric lymphoma (MALT lymphoma) · Linitis plasticaUpper and/or lowerAccessory exocrine pancreas: Adenocarcinoma · Pancreatic ductal carcinoma
cystic neoplasms: Serous microcystic adenoma · Intraductal papillary mucinous neoplasm · Mucinous cystic neoplasm · Solid pseudopapillary neoplasm
PancreatoblastomaPeritoneum Categories:- Hepatology
- Small blue round cell tumor
- Rare cancers
- Sarcoma
Wikimedia Foundation. 2010.