- DiGeorge syndrome
-
22q11.2 deletion syndrome Classification and external resources 
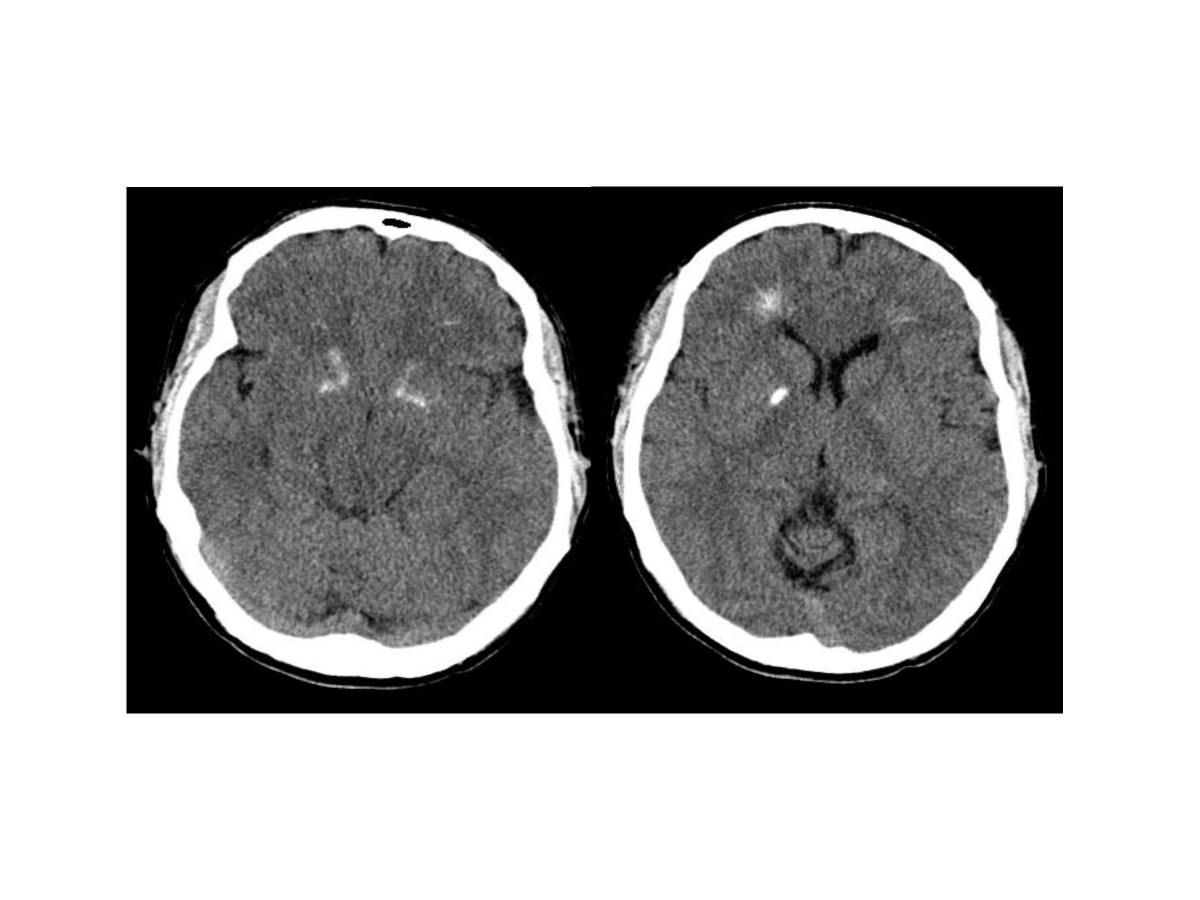
Brain computer tomography cuts of the patient, demonstrating basal ganglia and periventricular calcification. From a case report by Tonelli et al., 2007[1]ICD-10 D82.1 ICD-9 279.11, 758.32 OMIM 188400 DiseasesDB 3631 eMedicine med/567 ped/589 derm/716 MeSH D004062 22q11.2 deletion syndrome, which has several presentations including DiGeorge syndrome (DGS), DiGeorge anomaly,[2][3] velo-cardio-facial syndrome, Shprintzen syndrome, conotruncal anomaly face syndrome, Strong syndrome, congenital thymic aplasia, and thymic hypoplasia is a syndrome caused by the deletion of a small piece of chromosome 22. The deletion occurs near the middle of the chromosome at a location designated q11.2 i.e., on the long arm of one of the pair of chromosomes 22. It has a prevalence estimated at 1:4000.[4] The syndrome was described in 1968 by the pediatric endocrinologist Angelo DiGeorge.[5][6]
Contents
Presentation
The features of this syndrome vary widely, even among members of the same family, and affect many parts of the body. Characteristic signs and symptoms may include birth defects such as congenital heart disease, defects in the palate, most commonly related to neuromuscular problems with closure (velo-pharyngeal insufficiency), learning disabilities, mild differences in facial features, and recurrent infections. Infections are common in children due to problems with the immune system's T-cell mediated response that in some patients is due to an absent or hypoplastic thymus. 22q11.2 deletion syndrome may be first spotted when an affected newborn has heart defects or convulsions from hypocalcemia due to malfunctioning parathyroid glands and low levels of parathyroid hormone (parathormone). Affected individuals may also have any other kind of birth defect including kidney abnormalities and significant feeding difficulties as babies. Autoimmune disorders such as hypothyroidism and hypoparathyroidism or thrombocytopenia (low platelet levels), and psychiatric illnesses are common late-occurring features.[7] Microdeletions in chromosomal region 22q11.2 are associated with a 20 to 30-fold increased risk of schizophrenia.[8]
Studies provide various rates of 22q11.2 deletion syndrome in schizophrenia, ranging from 0.5 to 2% and averaging about 1%, compared with the overall estimated 0.025% risk of the 22q11.2 deletion syndrome in the general population.[9]
Salient features can be summarized using the mnemonic CATCH-22 to describe DiGeorge's syndrome, with the 22 to remind one the chromosomal abnormality is found on the 22 chromosome, as below:[10]
Cardiac Abnormality (especially tetralogy of Fallot)
Abnormal facies
Thymic aplasia
Cleft palate
Hypocalcemia.Nomenclature
The signs and symptoms of 22q11 deletion syndrome are so varied that different groupings of its features were once regarded as separate conditions. These original classifications included velo-cardio-facial syndrome, Shprintzen syndrome, DiGeorge sequence/syndrome, Sedlackova syndrome and conotruncal anomaly face syndrome. All are now understood to be presentations of a single syndrome.
Symptoms
Individuals with a 22q11.2 deletion can suffer from many possible features, ranging in number of associated features and from the mild to the very serious. Symptoms shown to be common include:
- Congenital heart disease (40% of individuals), particularly conotruncal malformations (tetralogy of Fallot, interrupted aortic arch, ventricular septal defect, and persistent truncus arteriosus)
- palatal abnormalities (50%), particularly velopharyngeal incompetence (VPI), submucosal cleft palate, and cleft palate; characteristic facial features (present in the majority of Caucasian individuals) including hypertelorism.
- learning difficulties (90%) but broad range
- hypocalcemia (50%)(due to hypoparathyroidism)
- significant feeding problems (30%)
- renal anomalies (37%)
- hearing loss (both conductive and sensorineural) (Hearing loss with craniofacial syndromes)
- laryngotracheoesophageal anomalies
- growth hormone deficiency
- autoimmune disorders
- seizures (without hypocalcemia)
- skeletal abnormalities
- psychiatric disorders
Cause
The syndrome is caused by genetic deletions (loss of a small part of the genetic material) found on the long arm of one of the two 22nd chromosomes. Very rarely, patients with somewhat similar clinical features may have deletions on the short arm of chromosome 10.
The mechanism that causes all of the associated features of the syndrome is unknown. 22q11.2 deletion syndrome may involve migration defects of neural crest-derived tissues, particularly affecting development of the third and fourth branchial pouches (pharyngeal pouches). This affects the thymus gland; a mediastinal organ largely responsible for differentiation and induction of tolerance in T-cells, and the Parathyroid glands, responsible for regulation of blood calcium levels.[11]
Treatment
There is no cure for 22q11.2 deletion syndrome. Certain individual features are treatable using standard treatments. The key is to identify each of the associated features and manage each using the best available treatments.
For example, in children it is important that the immune problems are identified early as special precautions are required regarding blood transfusion and immunisation with live vaccines. Thymus transplantation can be used to address absence of the thymus in the rare, so-called "complete" DiGeorge syndrome.[12] Bacterial infections are treated with antibiotics. Cardiac surgery is often required for congenital heart abnormalities. Hypoparathyroidism causing hypocalcaemia often requires lifelong vitamin D and calcium supplements.
Diagnosis and testing
The 22q11.2 deletion syndrome is diagnosed in individuals with a submicroscopic deletion of chromosome 22 detected by fluorescence in situ hybridization (FISH) or BACs-on-Beads technology. In FISH one DNA probes from the 22q11.2 chromosomal region is used at a time, while with BACs-on-Beads technology multiple probes from the 22q11.2 region can be used simultaneously. Such genetic testing is widely available for the clinical and prenatal testing of the 22q11.2 deletion syndrome. Fewer than 5% of individuals with clinical symptoms of the 22q11.2 deletion syndrome have normal routine cytogenetic studies and negative FISH testing. Some cases of DiGeorge Syndrome have defects in other chromosomes, notably a deletion in chromosome region 10p14. They may have variant deletions of DiGeorge syndrome that may be detectable on a research basis only or with other more advanced clinical testing method.
Genetics
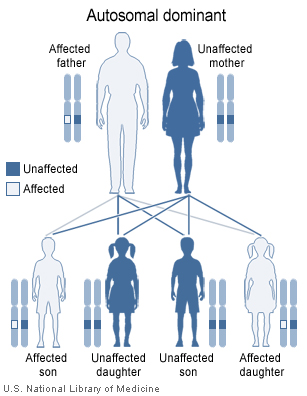
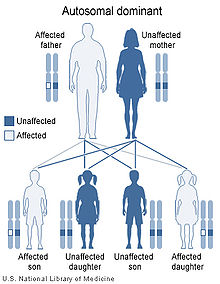 22q11.2 deletion syndrome is inherited in an autosomal dominant pattern.
22q11.2 deletion syndrome is inherited in an autosomal dominant pattern.
Most people with 22q11.2 deletion syndrome are missing about 3 million base pairs on one copy of chromosome 22 in every cell of their body. This region contains about 45 genes, but some of these genes have not been well characterized. A small percentage of affected individuals have shorter deletions in the same region.
Researchers have not yet identified the genes that contribute to the features of 22q11.2 deletion syndrome. They have determined that the loss of one particular gene on chromosome 22, TBX1, is probably responsible for some of the syndrome's characteristic signs (such as heart defects). Carrying only one copy of this gene does not appear to cause learning disabilities, however. Additional genes in the deleted region are likely to contribute to the signs and symptoms of 22q11.2 deletion syndrome and genes outside the 22q11.2 region may also play a role.
The 22q11.2 deletion syndrome can be inherited, but this is the case in the minority of newly diagnosed individuals. Only 5–10% have inherited the 22q11.2 deletion from a parent, whereas about 90–95% of cases have a de novo (new to the family) deletion of 22q11.2. This is because the 22q11.2 region has a structure that makes it highly prone to rearrangements during sperm or egg formation. The deletion is almost equally likely to occur when an egg is formed as when a sperm is formed. An individual with deletion 22q11.2 has a 50% (one in two) chance of passing the 22q11.2 deletion to their offspring. Prenatal testing, such as amniocentesis, is available for pregnancies determined to be at risk. Also, pregnancies with findings of congenital heart disease and/or palate anomalies detected by ultrasound examination may be offered prenatal testing for 22q11.2 deletion syndrome. Because most of the signs of this cluster of defects can also be inherited as autosomal recessive or x-linked traits, only genetic testing of both parents can determine with any certainty the likelihood of these anomalies occurring in any subsequent children.
Epidemiology
22q11.2 deletion syndrome affects between 1 in 2000 and 1 in 4000 live births.[4] This estimate is based on major birth defects and may be an underestimate, because some individuals with the deletion have few symptoms and may not have been formally diagnosed.
It is one of the most common causes of mental retardation due to a genetic deletion syndrome.[13]
Cognitive and language problems
Cognitive impairments
Children with 22q11.2 have a specific profile in neuropsychological tests. They usually have a borderline normal IQ with most individuals having higher scores in the verbal than the nonverbal domains.[contradictory] Cognitive functioning when processing information involving space and time usually shows significant impairment and this generally slows the development of numerical and arithmetical knowledge and skills.
Noteworthy is that these patients are a specifically high-risk group for developing schizophrenia. 30% have at least one incident of psychosis and about a quarter develop actual schizophrenia.[14]
Speech and language
Current research demonstrates there is a unique profile of speech and language impairments associated with 22q11.2 deletion syndrome. Children often perform lower on speech and language evaluations in comparison to their nonverbal IQ scores.[contradictory] Common problems include hypernasality, language delays, and speech sound errors.[15][16][17]
Hypernasality occurs when air escapes through the nose during the production of oral speech sounds resulting in reduced intelligibility. This is a common characteristic in the speech and language profile because 69% of children have palatal abnormalities. If the structure of the soft palate velum is such that it does not stop the flow of air from going up to the nasal cavity, it will cause hypernasal speech. This phenomenon is referred as velopharyngeal inadequacy VPI. Hearing loss can also contribute to increased hypernasality because children with hearing impairments can have difficulty self monitoring their oral speech output. The treatment options available for VPI include prosthesis and surgery.[15][16][18][19][20]
Difficulties acquiring vocabulary and formulating spoken language (expressive language deficits) at the onset of language development are also part of the speech and language profile associated with the 22q11.2 deletion. Vocabulary acquisition is often severely delayed for preschool age children. In some recent studies, children had a severely limited vocabulary or were still nonverbal at 2–3 years of age. School age children do make progress with expressive language as they mature, but many continue to have delays and demonstrate difficulty when presented with language tasks such as verbally recalling narratives and producing longer and more complex sentences. Receptive language, which is the ability to comprehend, retain, or process spoken language, can also be impaired although not usually with the same severity as expressive language impairments.[16][19][20][21]
Articulation errors are commonly present in children with 22q11.2 deletion syndrome. These errors include a limited phonemic (speech sound) inventory and the use of compensatory articulation strategies resulting in reduced intelligibility. The phonemic inventory typically produced consists of sounds made in the front or back of the vocal tract such as: /p/, /w/, /j/, /m/, /n/, and glottal stops. Mid vocal tract sounds are completely absent. Compensatory articulation errors made by this population of children include: glottal stops, nasal substitutions, pharyngeal fricatives, linguapalatal sibilants, reduced pressure on consonant sounds, or a combination of these symptoms. Of these errors, glottal stops have the highest frequency of occurrence. It is reasoned that a limited phonemic inventory and the use of compensatory articulation strategies is present due to the structural abnormalities of the palate. The speech impairments exhibited by this population are more severe during the younger ages and show a trend of gradual improvement as the child matures.[15][19]
See also
References
- ^ Tonelli AR, Kosuri K, Wei S, Chick D (2007). "Seizures as the first manifestation of chromosome 22q11.2 deletion syndrome in a 40-year old man: a case report". J Med Case Reports 1: 167. doi:10.1186/1752-1947-1-167. PMC 2222674. PMID 18053182. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2222674.
- ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ a b Oskarsdóttir S, Vujic M, Fasth A (2004). "Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden". Arch. Dis. Child. 89 (2): 148–51. doi:10.1136/adc.2003.026880. PMC 1719787. PMID 14736631. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1719787.
- ^ DiGeorge AM. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism. IV(1). White Plains, NY: March of Dimes-Birth Defects Foundation; 1968:116-21
- ^ Restivo, Angelo; Sarkozy, Anna; Digilio, Maria Cristina; Dallapiccola, Bruno; Marino, Bruno (February 2006). "22q11 Deletion syndrome: a review of some developmental biology aspects of the cardiovascular system.". Journal of Cardiovascular Medicine 7 (2): 77–85. doi:10.2459/01.JCM.0000203848.90267.3e. PMID 16645366. http://www.jcardiovascularmedicine.com/pt/re/jcm/abstract.01244665-200602000-00001.htm;jsessionid=K0GCjVnhWKskr2CxfrkZQlw2zDpw9lQTLJzcrSJFmNlGp7L8vQdK!713060492!181195629!8091!-1.
- ^ Debbané M, Glaser B, David MK, Feinstein C, Eliez S (2006). "Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: Neuropsychological and behavioral implications". Schizophr. Res. 84 (2–3): 187–93. doi:10.1016/j.schres.2006.01.019. PMID 16545541.
- ^ Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R (2003). "The schizophrenia phenotype in 22q11 deletion syndrome". Am J Psychiatry 160 (9): 1580–6. doi:10.1176/appi.ajp.160.9.1580. PMID 12944331.
- ^ Horowitz A, Shifman S, Rivlin N, Pisanté A, Darvasi A (2005). "A survey of the 22q11 microdeletion in a large cohort of schizophrenia patients". Schizophr. Res. 73 (2–3): 263–7. doi:10.1016/j.schres.2004.02.008. PMID 15653270.
- ^ Burn J (October 1999). "Closing time for CATCH22". J. Med. Genet. 36 (10): 737–8. PMC 1734243. PMID 10528851. http://jmg.bmj.com/cgi/pmidlookup?view=long&pmid=10528851.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) DiGeorge syndrome; DGS -188400
- ^ Markert ML, Devlin BH, Alexieff MJ (2007). "Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants". Blood 109 (10): 4539–47. doi:10.1182/blood-2006-10-048652. PMC 1885498. PMID 17284531. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1885498.
- ^ Daily DK, Ardinger HH, Holmes GE (February 2000). "Identification and evaluation of mental retardation". Am Fam Physician 61 (4): 1059–67, 1070. PMID 10706158. http://www.aafp.org/afp/20000215/1059.html.
- ^ Zinkstok J, van Amelsvoort T (2005). "Neuropsychological profile and neuroimaging in patients with 22Q11.2 Deletion Syndrome: a review". Child Neuropsychol 11 (1): 21–37. doi:10.1080/09297040590911194. PMID 15823981.
- ^ a b c D'Antonio LL, Scherer NJ, Miller LL, Kalbfleisch JH, Bartley JA (2001). "Analysis of speech characteristics in children with velocardiofacial syndrome (VCFS) and children with phenotypic overlap without VCFS". Cleft Palate Craniofac. J. 38 (5): 455–67. doi:10.1597/1545-1569(2001)038<0455:AOSCIC>2.0.CO;2. ISSN 1545-1569. PMID 11522167.
- ^ a b c Scherer NJ, D'Antonio LL, Kalbfleisch JH (1999). "Early speech and language development in children with velocardiofacial syndrome". Am. J. Med. Genet. 88 (6): 714–23. doi:10.1002/(SICI)1096-8628(19991215)88:6<714::AID-AJMG24>3.0.CO;2-B. PMID 10581495.
- ^ Scherer NJ, D'Antonio LL, Rodgers JR (2001). "Profiles of communication disorder in children with velocardiofacial syndrome: comparison to children with Down syndrome". Genet. Med. 3 (1): 72–8. doi:10.1097/00125817-200101000-00016. PMID 11339384.
- ^ Eliez S, Palacio-Espasa F, Spira A (2000). "Young children with Velo-Cardio-Facial syndrome (CATCH-22). Psychological and language phenotypes". Eur Child Adolesc Psychiatry 9 (2): 109–14. doi:10.1007/s007870050005. PMID 10926060.
- ^ a b c Robin NH, Shprintzen RJ (2005). "Defining the clinical spectrum of deletion 22q11.2". J. Pediatr. 147 (1): 90–6. doi:10.1016/j.jpeds.2005.03.007. PMID 16027702.
- ^ a b Solot CB, Knightly C, Handler SD (2000). "Communication disorders in the 22Q11.2 microdeletion syndrome". J Commun Disord 33 (3): 187–203; quiz 203–4. doi:10.1016/S0021-9924(00)00018-6. PMID 10907715.
- ^ Persson C, Niklasson L, Oskarsdóttir S, Johansson S, Jönsson R, Söderpalm E (2006). "Language skills in 5-8-year-old children with 22q11 deletion syndrome". Int J Lang Commun Disord 41 (3): 313–33. doi:10.1080/13682820500361497. PMID 16702096.
This article incorporates public domain text from The U.S. National Library of Medicine
External links
 Media related to DiGeorge Syndrome at Wikimedia Commons
Media related to DiGeorge Syndrome at Wikimedia Commons- DiGeorge syndrome at the Open Directory Project
- Velo-Cardio-Facial Syndrome Information Foundation
- McDonald-McGinn DM, Emanuel BS, Zackai EH (December 16, 2005). "22q11.2 Deletion Syndrome". In Pagon RA, Bird TD, Dolan CR, Stephens K. GeneReviews. PMID 20301696. NBK1523. http://www.ncbi.nlm.nih.gov/books/NBK1523/.
- Firth HV (February 17, 2009). "22q11.2 Duplication". In Pagon RA, Bird TD, Dolan CR, Stephens K. GeneReviews. PMID 20301749. NBK3823. http://www.ncbi.nlm.nih.gov/books/NBK3823/.
- Information on 22q11.2 Deletion syndrome from Seattle Children's Hospital
Immune disorders: Lymphoid and complement immunodeficiency (D80–D85, 279.0–4) Primary IgA deficiency · IgG deficiency · IgM deficiency · Hyper IgM syndrome (2, 3, 4, 5) · Wiskott-Aldrich syndrome · Hyper-IgE syndromeOtherthymic hypoplasia: hypoparathyroid (Di George's syndrome) · euparathyroid (Nezelof syndrome, Ataxia telangiectasia)x-linked: X-SCID
autosomal: Adenosine deaminase deficiency · Omenn syndrome · ZAP70 deficiency · Bare lymphocyte syndromeAcquired Leukopenia:
LymphocytopeniaComplement deficiency C1-inhibitor (Angioedema/Hereditary angioedema) · Complement 2 deficiency/Complement 4 deficiency · MBL deficiency · Properdin deficiency · Complement 3 deficiency · Terminal complement pathway deficiency · Paroxysmal nocturnal hemoglobinuria · Complement receptor deficiencyLymphatic disease: Congenital lymphatic organ disorders (Q89.0/Q89.2, 759.0/759.2) Thymus Thymic hypoplasia (DiGeorge syndrome) · Ectopic thymusSpleen Congenital asplenia/hyposplenism (Asplenia with cardiovascular anomalies) · Accessory spleen · PolyspleniaM: LMO
anat(h, u, t, a, l)/phys/depv
noco/cong/tumr
proc
Categories:- Autosomal dominant disorders
- IUIS-PID table 3 immunodeficiencies
- Noninfectious immunodeficiency-related cutaneous conditions
- Syndromes
- Autosomal monosomies and deletions
Wikimedia Foundation. 2010.