- B-cell chronic lymphocytic leukemia
-
"B-cell CLL" redirects here. For the gene family, see B-cell CLL/lymphoma.
Chronic lymphocytic leukemia Classification and external resources 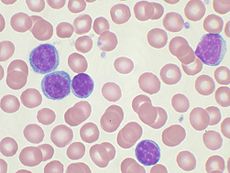
Peripheral blood smear showing CLL cellsICD-10 C91.1 ICD-9 204.1 ICD-O: M9823/3 (CLL)
9670/3 (SCL)DiseasesDB 2641 MedlinePlus 000532 eMedicine med/370 MeSH D015451 B-cell chronic lymphocytic leukemia (B-CLL), also known as chronic lymphoid leukemia (CLL), is the most common type of leukemia. Leukemias are cancers of the white blood cells (leukocytes). CLL affects B cell lymphocytes. B cells originate in the bone marrow, develop in the lymph nodes, and normally fight infection by producing antibodies. In CLL, the DNA of a B cell is damaged, so that it cannot produce antibodies.[citation needed] Additionally, B cells grow out of control and accumulate in the bone marrow and blood, where they crowd out healthy blood cells. CLL is a stage of small lymphocytic lymphoma (SLL), a type of B-cell lymphoma, which presents primarily in the lymph nodes.[1] CLL and SLL are considered the same underlying disease, just with different appearances.
CLL is a disease of adults, but, in rare cases, it can occur in teenagers and occasionally in children (inherited). Most (>75%) people newly diagnosed with CLL are over the age of 50, and the majority are men.
Most people are diagnosed without symptoms as the result of a routine blood test that returns a high white blood cell count, but, as it advances, CLL results in swollen lymph nodes, spleen, and liver, and eventually anemia and infections. Early CLL is not treated, and late CLL is treated with chemotherapy and monoclonal antibodies.
DNA analysis has distinguished two major types of CLL, with different survival times. CLL that is positive for the marker ZAP-70 has an average survival of 5 years. CLL that is negative for ZAP-70 has an average survival of more than 25 years. Patients with slowly-progressing disease can be reassured and may not need any treatment in their lifetimes.[2]
Contents
Symptoms and signs
Most people are diagnosed without symptoms as the result of a routine blood test that returns a high white blood cell count. Less commonly, CLL may present with enlarged lymph nodes without a high white blood cell count or no evidence of the disease in the blood. This is referred to as small lymphocytic lymphoma. In some individuals the disease comes to light only after the neoplastic cells overwhelm the bone marrow resulting in anemia producing tiredness or weakness.
Diagnosis
The disease is easily diagnosed. CLL is usually first suspected by the presence of a lymphocytosis, an increase in one type of white blood cell, on a complete blood count (CBC) test. This frequently is an incidental finding on a routine physician visit. Most often the lymphocyte count is greater than 4000 cells per microliter (µl) of blood, but can be much higher. The presence of a lymphocytosis in an elderly individual should raise strong suspicion for CLL, and a confirmatory diagnostic test, in particular flow cytometry, should be performed unless clinically unnecessary.
The diagnosis of CLL is based on the demonstration of an abnormal population of B lymphocytes in the blood, bone marrow, or tissues that display an unusual but characteristic pattern of molecules on the cell surface. This atypical molecular pattern includes the coexpression of cells surface markers cluster of differentiation 5 (CD5) and cluster of differentiation 23 (CD23). In addition, all the CLL cells within one individual are clonal, that is, genetically identical. In practice, this is inferred by the detection of only one of the mutually exclusive antibody light chains, kappa or lambda, on the entire population of the abnormal B cells. Normal B lymphocytes consist of a stew of different antibody-producing cells, resulting in a mixture of both kappa and lambda expressing cells. The lack of the normal distribution of kappa and lambda producing B cells is one basis for demonstrating clonality, the key element for establishing a diagnosis of any B cell malignancy (B cell non-Hodgkin lymphoma).
The combination of the microscopic examination of the peripheral blood and analysis of the lymphocytes by flow cytometry to confirm clonality and marker molecule expression is needed to establish the diagnosis of CLL. Both are easily accomplished on a small amount of blood. A flow cytometer is an instrument that can examine the expression of molecules on individual cells in fluids. This requires the use of specific antibodies to marker molecules with fluorescent tags recognized by the instrument. In CLL, the lymphocytes are genetically clonal, of the B cell lineage (express marker molecules cluster of differentiation 19 (CD19) and CD20), and characteristically express the marker molecules CD5 and CD23. These B cells resemble normal lymphocytes under the microscope, although slightly smaller, and are fragile when smeared onto a glass slide, giving rise to many broken cells (smudge cells).
Clinical staging
Staging, determining the extent of the disease, is done with the Rai staging system or the Binet classification (see details[3]) and is based primarily on the presence, or not, of a low platelet or red cell count. Early stage disease does not need to be treated.
Gene mutation status
Recent publications suggest that two[4] or three[5] prognostic groups of CLL exist based on the maturational state of the cell. This distinction is based on the maturity of the lymphocytes as discerned by the immunoglobulin variable-region heavy chain (IgVH) gene mutation status.[6] High risk patients have an immature cell pattern with few mutations in the DNA in the IgVH antibody gene region whereas low risk patients show considerable mutations of the DNA in the antibody gene region indicating mature lymphocytes.[7]
Since assessment of the IgVH antibody DNA changes is difficult to perform, the presence of either cluster of differentiation 38 (CD38) or Z-chain–associated protein kinase-70 (ZAP-70) may be surrogate markers of high risk subtype of CLL.[6] Their expression correlates with a more immature cellular state and a more rapid disease course.
Fluorescence in situ hybridization
In addition to the maturational state, the prognosis of patients with CLL is dependent on the genetic changes within the neoplastic cell population. These genetic changes can be identified by fluorescent probes to chromosomal parts using a technique referred to as fluorescent in situ hybridization (FISH).[6] Four main genetic aberrations are recognized in CLL cells that have a major impact on disease behavior.[8]
- Deletions of part of the short arm of chromosome 17 (del 17p), which target the cell cycle regulating protein p53 are particularly deleterious. Patients with this abnormality have significantly short interval before they require therapy and a shorter survival. This abnormality is found in 5–10% of patients with CLL.
- Deletions of the long arm on chromosome 11 (del 11q) are also unfavorable although not to the degree seen with del 17p. The abnormality targets the ATM gene and occurs infrequently in CLL (5–10%).
- Trisomy 12, an additional chromosome 12, is a relatively frequent finding occurring in 20–25% of patients and imparts an intermediate prognosis.
- Deletion of the long arm of chromosome 13 (del 13q) is the most common abnormality in CLL with roughly 50% of patients with cells containing this defect. These patients have the best prognosis and most will live many years, even decades, without the need for therapy. The gene targeted by this deletion is a segment coding for microRNAs miR-15a and miR-16-1.
Array-based karyotyping
Array-based karyotyping is a cost-effective alternative to FISH for detecting chromosomal abnormalities in CLL. Several clinical validation studies have shown >95% concordance with the standard CLL FISH panel.[9][10][11][12][13] See also Virtual Karyotype.
Related diseases
In the past, cases with similar microscopic appearance in the blood but with a T cell phenotype were referred to as T-cell CLL. These so-called T-cell CLLs, however, are now recognized as a separate disease group and are currently classified as T-cell prolymphocytic leukemias.[14][15]
CLL should not be confused with acute lymphoblastic leukemia (ALL), a highly aggressive and highly treatable leukemia most commonly diagnosed in children.
Differential diagnosis
Hematologic disorders that may resemble CLL in their clinical presentation, behavior, and microscopic appearance include mantle cell lymphoma, marginal zone lymphoma, B cell prolymphocytic leukemia, and lymphoplasmacytic lymphoma.
- B cell prolymphocytic leukemia (B PLL), a related, but more aggressive disorder, has cells with similar phenotype, but are significantly larger than normal lymphocytes and have a prominent nucleolus. The distinction is important as the prognosis and therapy differs from CLL.
- Hairy cell leukemia is also a neoplasm of B lymphocytes, but the neoplastic cells have a distinct morphology under the microscope (hairy cell leukemia cells have delicate, hair-like projections on their surface) and unique marker molecule expression.
All the B cell malignancies of the blood and bone marrow can be differentiated from one another by the combination of cellular microscopic morphology, marker molecule expression, and specific tumor-associated gene defects. This is best accomplished by evaluation of the patient's blood, bone marrow and occasionally lymph node cells by a pathologist with specific training in blood disorders. A flow cytometer is necessary for cell marker analysis, and the detection of genetic problems in the cells may require visualizing the DNA changes with fluorescent probes by FISH.
Treatment
CLL treatment focuses on controlling the disease and its symptoms rather than on an outright cure. CLL is treated by chemotherapy, radiation therapy, biological therapy, or bone marrow transplantation. Symptoms are sometimes treated surgically (splenectomy removal of enlarged spleen) or by radiation therapy ("de-bulking" swollen lymph nodes).
Initial CLL treatments vary depending on the exact diagnosis and the progression of the disease, and even with the preference and experience of the health care practitioner. There are dozens of agents used for CLL therapy.[16]
Decision to treat
While generally considered incurable, CLL progresses slowly in most cases. Many people with CLL lead normal and active lives for many years—in some cases for decades. Because of its slow onset, early-stage CLL is, in general, not treated since it is believed that early CLL intervention does not improve survival time or quality of life. Instead, the condition is monitored over time to detect any change in the disease pattern.
The decision to start CLL treatment is taken when the patient's clinical symptoms or blood counts indicate that the disease has progressed to a point where it may affect the patient's quality of life.
Clinical "staging systems" such as the Rai 4-stage system and the Binet classification can help to determine when and how to treat the patient.[3]
Determining when to start treatment and by what means is often difficult; studies have shown there is no survival advantage to treating the disease too early. The National Cancer Institute Working Group has issued guidelines for treatment, with specific markers that should be met before it is initiated.[17]
Chemotherapy
Combination chemotherapy regimens are effective in both newly-diagnosed and relapsed CLL. Combinations of fludarabine with alkylating agents (cyclophosphamide) produce higher response rates and a longer progression-free survival than single agents:
- FC (fludarabine with cyclophosphamide)[18]
- FR (fludarabine with rituximab)[19]
- FCR (fludarabine, cyclophosphamide, and rituximab)[20]
- CHOP (cyclophosphamide, doxorubicin, vincristine and prednisolone)
Although the purine analogue fludarabine was shown to give superior response rates to chlorambucil as primary therapy,[21][22] there is no evidence early use of fludarabine improves overall survival, and some clinicians prefer to reserve fludarabine for relapsed disease.
Alkylating agents approved for CLL include bendamustine and cyclophosphamide.
Monoclonal antibodies, such as alemtuzumab (directed against CD52) and rituximab and ofatumumab (directed against CD20), are also used.
Stem cell transplantation
Allogeneic bone marrow (stem cell) transplantation is rarely used as a first-line treatment for CLL due to its risk. There is increasing interest in the use of reduced-intensity allogeneic stem cell transplantation, which offers the prospect of cure for selected patients with a suitable donor.[23]
Younger patients that are at high risk for dying from CLL might consider hematopoietic stem cell transplantation (HSCT). Autologous stem cell transplantation, a lower-risk form of treatment using the patient's own blood cells, is not curative. Myeloablative (bone marrow killing) forms of allogeneic stem cell transplantation, a high-risk treatment using blood cells from a healthy donor, may be curative for some patients, but most patients cannot tolerate the treatment. An intermediate level, called reduced-intensity conditioning allogeneic stem cell transplantation, may be better tolerated by older or frail patients.[24]
Refractory CLL
"Refractory" CLL is a disease that no longer responds favorably to treatment. In this case more aggressive therapies, including lenalidomide, flavopiridol, and bone marrow (stem cell) transplantation, are considered.[25] The monoclonal antibody, alemtuzumab (directed against CD52), may be used in patients with refractory, bone marrow-based disease.[26]
Complications
Chronic lymphocytic leukemia may transform into Richter's syndrome, a term used to describe the development of fast-growing diffuse large B cell lymphoma, prolymphocytic leukemia, Hodgkin's lymphoma, or acute leukemia in a patient who has chronic lymphocytic leukemia. Its incidence is estimated to be around 5%.[27]
Gastrointestinal (GI) involvement can rarely occur with chronic lymphocytic leukemia. Some of the reported manifestations include intussusception, small intestinal bacterial contamination, colitis and others. Usually, GI complications with CLL occur after Richter transformation. There have been two case reports to date of GI involvement in chronic lymphocytic leukemia without Richter's transformation.[28]
Epidemiology
CLL is a disease of older adults and is rarely encountered in individuals under the age of 40. Thereafter, the disease incidence increases with age.
In the United States during 2009, about 16,000 new cases are expected to be diagnosed, and 4,400 patients are expected to die from CLL.[3] Because of the prolonged survival, which was typically about ten years in past decades, but which can extend to a normal life expectancy,[3] the prevalence (number of people living with the disease) is much higher than the incidence (new diagnoses).
Subclinical "disease" can be identified in 3.5% of normal adults,[29] and in up to 8% of individuals over the age of 70[citation needed]. That is, small clones of B cells with the characteristic CLL phenotype can be identified in many healthy elderly persons. The clinical significance of these cells is unknown.
Of all cancers involving the same class of blood cell, 7% of cases are CLL/SLL.[30]
Complications include hypogammaglobulinemia leading to recurrent infection, warm autoimmune hemolytic anaemia in 10–15% of patients, transformation to high grade lymphoma, and Richter's transformation.
Rates of CLL are somewhat elevated in people exposed to certain chemicals. Under U.S. Department of Veterans' Affairs regulations, Vietnam veterans who served in-country or in the inland waterways of Vietnam and who later develop CLL are presumed to have contracted it from exposure to Agent Orange and may be entitled to compensation.[31]
Prognosis
Prognosis depends on the subtype. The overall five-year survival rate (all forms of CLL together) is about 50%.[32]
Research directions
Considerable research activity, studying the many treatments individually or in combination, is ongoing.[16] Current research is comparing different forms of bone marrow transplants to determine which patients are the best candidates and which approach is best in different situations.[24]
Researchers at the Abramson Cancer Center of the University of Pennsylvania School of Medicine reported preliminary success in the use of gene therapy, through genetically modified T cells, to treat CLL.[33] The findings, which were published in August 2011,[34][35] were based on data from three patients who had modified T cells injected into their blood. The T cells had been modified to express genes that would allow the cells to proliferate in the body and destroy B cells including those causing the leukemia. Two patients went into remission, while the presence of leukemia in the third patient reduced by 70 percent.[36][37] One of the patients had been diagnosed with CLL for 13 years, and his treatment was failing before he participated in the clinical trial. One week after the T cells were injected, the leukemia cells in his blood had disappeared.[38] The T cells were still found in the bloodstream of the patients six months after the procedure, meaning they would be able to fight the disease should leukemia cells return.[36] This was the first time scientists "have used gene therapy to successfully destroy cancer tumors in patients with advanced disease".[39] One editorial in The New England Journal of Medicine also urged caution because only more trials would indicate whether the findings are "an authentic advance toward a clinically applicable and effective therapy".[40][41]
In pregnancy
Leukemia is rarely associated with pregnancy, affecting only about 1 in 10,000 pregnant women.[42] Treatment for chronic lymphocytic leukemias can often be postponed until after the end of the pregnancy. If treatment is necessary, then giving chemotherapy during the second or third trimesters is less likely to result in pregnancy loss or birth defects than treatment during the first trimester.[42]
See also
- Monoclonal B-cell lymphocytosis
- Virtual Karyotype
- B-cell CLL/lymphoma
References
- ^ Harris NL, Jaffe ES, Diebold J et al. (1999). "World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997". J. Clin. Oncol. 17 (12): 3835–49. PMID 10577857.
- ^ Chiorazzi N, Rai KR, Ferrarini M (2005). "Chronic lymphocytic leukemia". N. Engl. J. Med. 352 (8): 804–15. doi:10.1056/NEJMra041720. PMID 15728813.
- ^ a b c d National Cancer Institute. "Chronic Lymphocytic Leukemia (PDQ) Treatment: Stage Information". http://www.cancer.gov/cancertopics/pdq/treatment/CLL/HealthProfessional/page2. Retrieved 2007-09-04.
- ^ Rosenwald A, Alizadeh AA, Widhopf G et al. (2001). "Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia". J. Exp. Med. 194 (11): 1639–47. doi:10.1084/jem.194.11.1639. PMC 2193523. PMID 11733578. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2193523.
- ^ Ghia P, Guida G, Stella S et al. (2003). "The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression". Blood 101 (4): 1262–9. doi:10.1182/blood-2002-06-1801. PMID 12406914.
- ^ a b c Shanafelt TD, Byrd JC, Call TG, Zent CS, Kay NE (2006). "Narrative review: initial management of newly diagnosed, early-stage chronic lymphocytic leukemia". Ann. Intern. Med. 145 (6): 435–47. PMID 16983131. http://www.annals.org/cgi/content/full/145/6/435.
- ^ Hamblin TJ, Davis Z, Gardiner A et al. (1999). "Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia". Blood 94 (6): 1848–54. PMID 10477713. http://bloodjournal.hematologylibrary.org/cgi/content/full/94/6/1848.
- ^ Dohner H, Stilgenbauer S, Benner A, "" et al. (2000). "Genomic aberrations and survival in chronic lymphocytic leukemia". NEJM 343 (26): 1910–6. doi:10.1056/NEJM200012283432602. PMID 11136261. http://content.nejm.org/cgi/content/full/343/26/1910.
- ^ Lehmann S, Ogawa S, Raynaud SD et al. (March 2008). "Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia". Cancer 112 (6): 1296–305. doi:10.1002/cncr.23270. PMID 18246537.
- ^ Sargent R, Jones D, Abruzzo LV et al. (January 2009). "Customized oligonucleotide array-based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia". J Mol Diagn 11 (1): 25–34. doi:10.2353/jmoldx.2009.080037. PMC 2607562. PMID 19074592. http://linkinghub.elsevier.com/retrieve/pii/S1525-1578(10)60205-X.
- ^ Schwaenen C, Nessling M, Wessendorf S et al. (January 2004). "Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations". Proc. Natl. Acad. Sci. U.S.A. 101 (4): 1039–44. doi:10.1073/pnas.0304717101. PMC 327147. PMID 14730057. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=14730057.
- ^ Pfeifer D, Pantic M, Skatulla I et al. (February 2007). "Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays". Blood 109 (3): 1202–10. doi:10.1182/blood-2006-07-034256. PMID 17053054. http://www.bloodjournal.org/cgi/pmidlookup?view=long&pmid=17053054.
- ^ Gunn SR, Mohammed MS, Gorre ME et al. (September 2008). "Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia". J Mol Diagn 10 (5): 442–51. doi:10.2353/jmoldx.2008.080033. PMC 2518739. PMID 18687794. http://linkinghub.elsevier.com/retrieve/pii/S1525-1578(10)60182-1.
- ^ "T Cell Prolymphocytic Leukemia". AccessMedicine. http://www.accessmedicine.com/content.aspx?aid=2149838. Retrieved 2009-02-04.
- ^ Ascani S, Leoni P, Fraternali Orcioni G et al. (June 1999). "T-cell prolymphocytic leukaemia: does the expression of CD8+ phenotype justify the identification of a new subtype? Description of two cases and review of the literature". Ann. Oncol. 10 (6): 649–53. doi:10.1023/A:1008349422735. PMID 10442186. http://annonc.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=10442186.
- ^ a b National Cancer Institute. "Chronic Lymphocytic Leukemia (PDQ) Treatment: Stage I, II, III, and IV Chronic Lymphocytic Leukemia". http://www.cancer.gov/cancertopics/pdq/treatment/CLL/HealthProfessional/page5. Retrieved 2007-09-04.
- ^ Cheson BD, Bennett JM, Grever M et al. (1996). "National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment". Blood 87 (12): 4990–7. PMID 8652811.
- ^ Eichhorst BF, Busch R, Hopfinger G, Pasold R, Hensel M, Steinbrecher C, Siehl S, Jäger U, Bergmann M, Stilgenbauer S, Schweighofer C, Wendtner CM, Döhner H, Brittinger G, Emmerich B, Hallek M, German CLL Study Group. (2006). "Fludarabine plus cyclophosphamide versus fludarabine alone in first-line therapy of younger patients with chronic lymphocytic leukemia". Blood 107 (3): 885–91. doi:10.1182/blood-2005-06-2395. PMID 16219797.
- ^ Byrd JC, Peterson BL, Morrison VA et al. (2003). "Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712)". Blood 101 (1): 6–14. doi:10.1182/blood-2002-04-1258. PMID 12393429.
- ^ Keating MJ, O'Brien S, Albitar M et al. (2005). "Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia". J. Clin. Oncol. 23 (18): 4079–88. doi:10.1200/JCO.2005.12.051. PMID 15767648.
- ^ Rai KR, Peterson BL, Appelbaum FR et al. (2000). "Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia". N. Engl. J. Med. 343 (24): 1750–7. doi:10.1056/NEJM200012143432402. PMID 11114313.
- ^ Steurer M, Pall G, Richards S, Schwarzer G, Bohlius J, Greil R (2006). Steurer, Michael. ed. "Purine antagonists for chronic lymphocytic leukaemia". Cochrane database of systematic reviews (Online) 3: CD004270. doi:10.1002/14651858.CD004270.pub2. PMID 16856041.
- ^ Dreger P, Brand R, Hansz J, Milligan D, Corradini P, Finke J, Deliliers GL, Martino R, Russell N, Van Biezen A, Michallet M, Niederwieser D; Chronic Leukemia Working Party of the EBMT (2003). "Treatment-related mortality and graft-versus-leukemia activity after allogeneic stem cell transplantation for chronic lymphocytic leukemia using intensity-reduced conditioning". Leukemia 17 (5): 841–8. doi:10.1038/sj.leu.2402905. PMID 12750695.
- ^ a b Gribben JG (January 2008). "Stem cell transplantation in chronic lymphocytic leukemia". Biol. Blood Marrow Transplant. 15 (1 Suppl): 53–8. doi:10.1016/j.bbmt.2008.10.022. PMC 2668540. PMID 19147079. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2668540.
- ^ National Cancer Institute. "Chronic Lymphocytic Leukemia (PDQ) Treatment: Refractory Chronic Lymphocytic Leukemia". http://www.cancer.gov/cancertopics/pdq/treatment/CLL/HealthProfessional/page6. Retrieved 2007-09-04.
- ^ Keating MJ, Flinn I, Jain V, Binet JL, Hillmen P, Byrd J, Albitar M, Brettman L, Santabarbara P, Wacker B, Rai KR (2002). "Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study". Blood 99 (10): 3554–61. doi:10.1182/blood.V99.10.3554. PMID 11986207.
- ^ Tsimberidou AM, Keating MJ (January 2005). "Richter syndrome: biology, incidence, and therapeutic strategies". Cancer 103 (2): 216–28. doi:10.1002/cncr.20773. PMID 15578683.
- ^ Bitetto AM, Lamba G, Cadavid G, Shah D, Forlenza T, Rotatori F, Rafiyath SM. Colonic perforation secondary to chronic lymphocytic leukemia infiltration without Richter transformation. Leuk Lymphoma. 2011 May;52(5):930-3.
- ^ Rawstron AC, Green MJ, Kuzmicki A et al. (July 2002). "Monoclonal B lymphocytes with the characteristics of "indolent" chronic lymphocytic leukemia are present in 3.5% of adults with normal blood counts". Blood 100 (2): 635–9. doi:10.1182/blood.V100.2.635. PMID 12091358. http://www.bloodjournal.org/cgi/pmidlookup?view=long&pmid=12091358.
- ^ Turgeon, Mary Louise (2005). Clinical hematology: theory and procedures. Hagerstown, MD: Lippincott Williams & Wilkins. p. 283. ISBN 0-7817-5007-5. "Frequency of lymphoid neoplasms. (Source: Modified from WHO Blue Book on Tumour of Hematopoietic and Lymphoid Tissues. 2001, p. 2001.)"
- ^ http://ecfr.gpoaccess.gov/cgi/t/text/text-idx?c=ecfr&sid=150fea5ac37ad66e06bdb4f940a0e0dc&rgn=div8&view=text&node=38:1.0.1.1.4.1.65.117&idno=38>
- ^ "Chronic Leukemias". Merck Manual of Geriatrics. http://www.merck.com/mkgr/mmg/sec9/ch73/ch73b.jsp.
- ^ Auer, Holly (August 10, 2011). "Genetically Modified "Serial Killer" T Cells Obliterate Tumors in Patients with Chronic Lymphocytic Leukemia, Penn Researchers Report". University of Pennsylvania School of Medicine. http://www.uphs.upenn.edu/news/News_Releases/2011/08/t-cells/. Retrieved August 12, 2011.
- ^ Porter DL et al. (2011). "Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia". N. Engl. J. Med.: 110810110014063. doi:10.1056/NEJMoa1103849. PMID 21830940.
- ^ Kalos M et al. (2011). "T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia". Sci. Transl. Med. 3 (95): 95ra73. doi:10.1126/scitranslmed.3002842. PMID 21832238.
- ^ a b Palca, Joe (August 11, 2011). "Gene Therapy Advance Trains Immune System To Fight Leukemia". NPR. http://www.npr.org/blogs/health/2011/08/11/139536661/gene-therapy-breakthrough-trains-immune-system-to-fight-leukemia. Retrieved August 12, 2011.
- ^ Bazell, Robert (August 10, 2011). "New leukemia treatment exceeds 'wildest expectations'". MSNBC. http://www.msnbc.msn.com/id/44090512/. Retrieved August 12, 2011.
- ^ DeNoon, Daniel J. (August 10, 2011). "Gene Therapy Cures Adult Leukemia". WebMD. http://www.webmd.com/cancer/news/20110810/gene-therapy-cures-adult-leukemia. Retrieved August 12, 2011.
- ^ Beasly, Deena (August 10, 2011). "Gene therapy shown to destroy leukemia tumors". Reuters. http://www.reuters.com/article/2011/08/10/us-leukemia-genetherapy-idUSTRE7795NT20110810. Retrieved August 12, 2011.
- ^ "Treatment for Leukemia Is Showing Early Promise". The New York Times. Associated Press: p. A15. August 11, 2011. http://www.nytimes.com/2011/08/11/health/research/11cancer.html. Retrieved August 12, 2011.
- ^ Urba WJ et al. (2011). "Redirecting T Cells". N. Engl. J. Med.: 110810110014063. doi:10.1056/NEJMe1106965. PMID 21830939.
- ^ a b Shapira T, Pereg D, Lishner M (September 2008). "How I treat acute and chronic leukemia in pregnancy". Blood Rev. 22 (5): 247–59. doi:10.1016/j.blre.2008.03.006. PMID 18472198.
External links
- Chronic Lymphocytic Leukemia at American Cancer Society
- CLL booklet from Leukemia & Lymphoma Society
- General information about CLL from the US National Cancer Institute
Hematological malignancy/leukemia histology (ICD-O 9590–9989, C81–C96, 200–208)
Lymphoid/Lymphoproliferative, Lymphomas/Lymphoid leukemias (9590–9739, 9800–9839)By development/
markerALL (Precursor B acute lymphoblastic leukemia/lymphoma)CD5+naive B cell (CLL/SLL)CD22+germinal center/follicular B cell (Follicular, Burkitt's, GCB DLBCL, Primary cutaneous follicular lymphoma)marginal zone/marginal-zone B cell (Splenic marginal zone, MALT, Nodal marginal zone, Primary cutaneous marginal zone lymphoma)see immunoproliferative immunoglobulin disordersBy infectionDiffuse large B-cell lymphoma · Intravascular large B-cell lymphoma · Primary cutaneous marginal zone lymphoma · Primary cutaneous immunocytoma · Plasmacytoma · Plasmacytosis · Primary cutaneous follicular lymphomaBy development/
markerTdT+: ALL (Precursor T acute lymphoblastic leukemia/lymphoma)
prolymphocyte (Prolymphocytic)
CD30+ (Anaplastic large-cell lymphoma, Lymphomatoid papulosis type A)indolent: Mycosis fungoides · Pagetoid reticulosis · Granulomatous slack skin
aggressive: Sézary's disease · Adult T-cell leukemia/lymphomaNon-MFCD30-: Non-mycosis fungoides CD30− cutaneous large T-cell lymphoma · Pleomorphic T-cell lymphoma · Lymphomatoid papulosis type B
CD30+: CD30+ cutaneous T-cell lymphoma · Secondary cutaneous CD30+ large cell lymphoma · Lymphomatoid papulosis type AOther peripheralHepatosplenic · Angioimmunoblastic · Enteropathy-associated T-cell lymphoma · Peripheral T-cell lymphoma-Not-Otherwise-Specified (Lennert lymphoma) · Subcutaneous T-cell lymphomaBy infectionHTLV-1 (Adult T-cell leukemia/lymphoma)NK cell/
(most CD56)Aggressive NK-cell leukemia · Blastic NK cell lymphomaT or NKLymphoid+myeloidCutaneous lymphoid hyperplasia Cutaneous lymphoid hyperplasia with bandlike and perivascular patterns · Cutaneous lymphoid hyperplasia with nodular pattern · Jessner lymphocytic infiltrate of the skinB-cell chronic lymphocytic leukemia · Hepatoblastoma · Rhabdomyosarcoma · Ewing's sarcoma · Neuroblastoma · Retinoblastoma · Wilms' tumorCategories:- Lymphocytic leukemia
- Small blue round cell tumor
Wikimedia Foundation. 2010.