- Organophosphorus
-
Organophosphorus compounds are degradable organic compounds containing carbon–phosphorus bonds (thus excluding from phosphate and phosphite esters, which lack such kind of bonding), used primarily in pest control as an alternative to chlorinated hydrocarbons that persist in the environment. Organophosphorus chemistry is the corresponding science of the properties and reactivity of organophosphorus compounds. Phosphorus shares group 5 in the periodic table with nitrogen and phosphorus compounds and nitrogen compounds are somewhat related.[1][2][3]
The definition of organophosphorus compounds is variable, which can lead to confusion. In industrial and environmental chemistry, an organophosphorus compound need contain only an organic substituent, but need not have a direct P-C bond. Thus most pesticides, e.g., malathion, are often included in this class of compounds.
Phosphorus can adopt a variety of oxidation states, and it is general to classify organophosphorus compounds based on their being derivatives of phosphorus(V) vs phosphorus(III), which are the predominant classes of compounds. In a descriptive but only intermittently used nomenclature, phosphorus compounds are identified by their coordination number δ and their valency λ. In this system, a phosphine is a δ3λ3 compound.
Contents
Organophosphorus(V) compounds, main categories
Phosphate esters and amides
Main article: OrganophosphatePhosphate esters have the general structure P(=O)(OR)3 feature P(V). Such species are of technological importance as flame retardant agents, and plasticizers. Lacking a P−C bond, these compounds are in the technical sense not organophosphorus compounds but esters of phosphoric acid. Many derivatives are found in nature, such as phosphatidylcholine. Phosphate ester are synthesized by alcoholysis of phosphorus oxychloride. A variety of mixed amido-alkoxo derivatives are known, one medically significant example being the anti-cancer drug cyclophosphamide. Also derivatives containing the thiophosphoryl group (P=S) include the pesticide malathion. The organophosphates prepared on the largest scale are the zinc dithiophosphates, as additives for motor oil. Several million kilograms of this coordination complex are produced annually by the reaction of phosphorus pentasulfide with alcohols.[4]
-
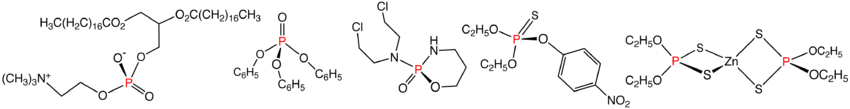 Illustrative organophosphates and related compounds: phosphatidylcholine, triphenylphosphate, cyclophosphamide, parathion, and zinc dithiophosphate.
Illustrative organophosphates and related compounds: phosphatidylcholine, triphenylphosphate, cyclophosphamide, parathion, and zinc dithiophosphate.
In the environment, these compounds break down via hydrolysis to eventually afford phosphate and the organic alcohol or amine from which they are derived.
Phosphonic and phosphinic acids and their esters
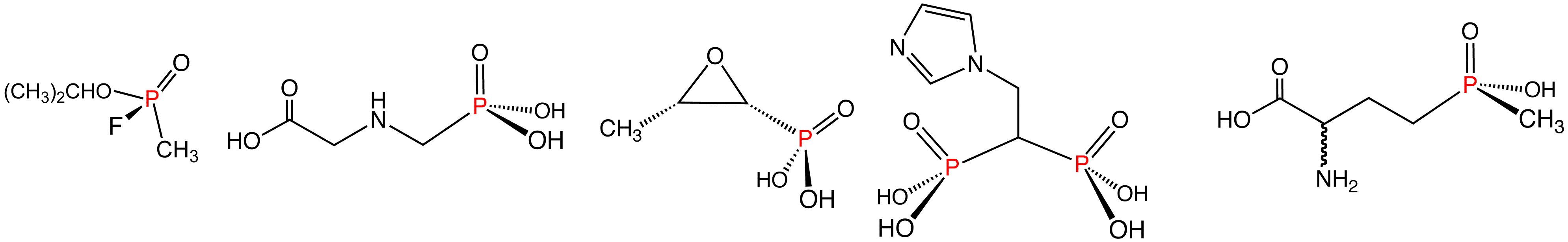
Main articles: Phosphonate and PhosphinatePhosphonates are esters of phosphonic acid and have the general formula RP(=O)(OR')2. Phosphonates have many technical applications, a well-known member being Glyphosate, better known as Roundup. With the formula (HO)2P(O)CH2NHCH2CO2H, this derivative of glycine is one of the most widely used herbicides. Bisphosphonates are a class of drugs to treat osteoporosis. The nerve gas agent Sarin, containing both C–P and F–P bonds, is a phosphonate.
Phosphinates feature two P–C bonds, with the general formula R2P(=O)(OR'). A commercially significant member is the herbicide Glufosinate. Similar to Glyphosate mentioned above, it has the structure CH3P(O)(OH)CH2CH2CH(NH2)CO2H.
-
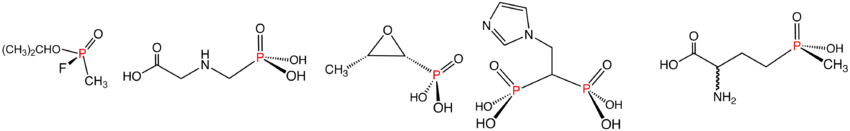 Illustrative examples of phosphonates and phosphinates in the order shown: Sarin (phosphinate), Glyphosate (phosphonate), fosfomycin (phosphonate), zoledronic acid (phosphonate), and Glufosinate (phosphinate). In aqueous solution, phosphonic acids ionize to give the corresponding organophosphonates.
Illustrative examples of phosphonates and phosphinates in the order shown: Sarin (phosphinate), Glyphosate (phosphonate), fosfomycin (phosphonate), zoledronic acid (phosphonate), and Glufosinate (phosphinate). In aqueous solution, phosphonic acids ionize to give the corresponding organophosphonates.
The Michaelis–Arbuzov reaction is the main method for the synthesis of these compounds. For example, dimethylmethylphosphonate (see figure above) arises from the rearrangement of trimethylphosphite, which is catalyzed by methyl iodide. In the Horner–Wadsworth–Emmons reaction and the Seyferth–Gilbert homologation, phosphonates are used in reactions with carbonyl compounds. The Kabachnik–Fields reaction is a method for the preparation of aminophosphonates. These compounds contain a very inert bond between phosphorus and carbon. Consequently, they hydrolyze to give phosphonic and phosphinic acid derivatives, but not phosphate.
Phosphine oxides, imides, and chalcogenides
Main article: Phosphine oxidePhosphine oxides (designation δ3λ3) have the general structure R3P=O with formal oxidation state V. Phosphine oxides form hydrogen bonds and some are therefore soluble in water. The P=O bond is very polar with a dipole moment of 4.51 D for triphenylphosphine oxide.
Compounds related to phosphine oxides are the imides (R3PNR') and related chalcogenides (R3PE, where E = S, Se, Te). These compounds are some of the most thermally stable organophosphorus compounds, but few are useful in significant amounts.
Phosphonium salts and phosphoranes
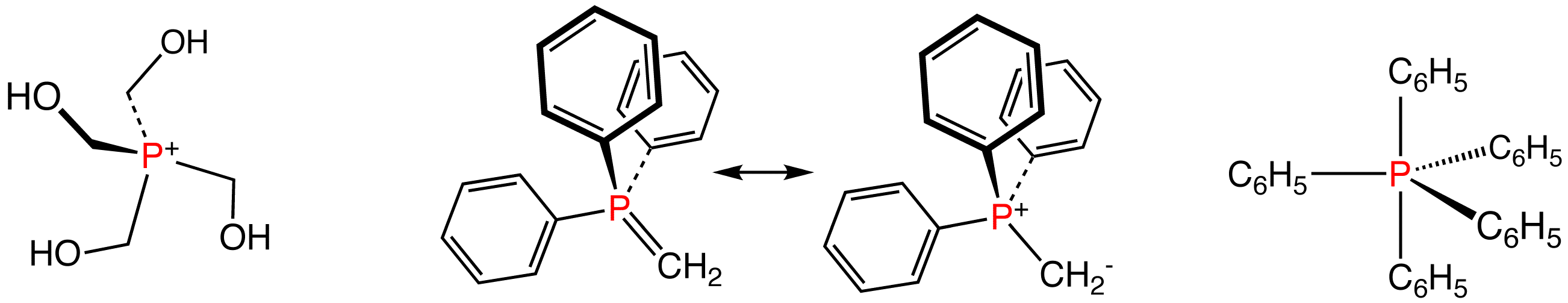
Compounds with the formula [PR4+]X- comprise the phosphonium salts. These species are tetrahedral phosphorus(V) compounds. From the commercial perspective, the most important member is tetrakis(hydroxymethyl)phosphonium chloride, [P(CH2OH)4]Cl, which is used as a fire retardant in textiles. Approximately 2M kg are produced annually of the chloride and the related sulfate.[4] They are generated by the reaction of phosphine with formaldehyde in the presence of the mineral acid:
- PH3 + HX + 4 CH2O → [P(CH2OH)4+]X-
A variety of phosphonium salts can be prepared by alkylation and arylation of organophosphines:
- PR3 + R'X → [PR3R'+]X-
The methylation of triphenylphosphine is the first step in the preparation of the Wittig reagent.
-
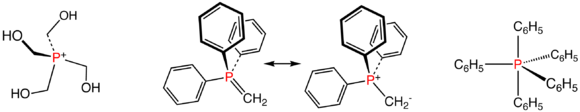 Illustrative phosphorus(V) compounds: the phosphonium ion P(CH2OH)4+, two resonance structures for the Wittig reagent Ph3PCH2, and pentaphenylphosphorane, a rare pentaorganophophorus compound.
Illustrative phosphorus(V) compounds: the phosphonium ion P(CH2OH)4+, two resonance structures for the Wittig reagent Ph3PCH2, and pentaphenylphosphorane, a rare pentaorganophophorus compound.
The parent phosphorane (δ5λ5) is PH5, which is unknown. Related compounds containing both halide and organic substituents on phosphorus are fairly common. Those with five organic substituents are rare, although P(C6H5)5 is known, being derived from P(C6H5)4+ by reaction with phenyllithium.
Phosphorus ylides are unsaturated phosphoranes, known as Wittig reagents, e.g. CH2P(C6H5)3. These compounds feature tetrahedral phosphorus(V) and are considered relatives of phosphine oxides. They also are derived from phosphonium salts, but by deprotonation not alkylation.
Organophosphorus(III) compounds, main categories
Phosphites, phosphonites, and phosphinites
Phosphites, sometimes called phosphite esters, have the general structure P(OR)3 with oxidation state +3. Such species arise from the alcoholysis of phosphorus trichloride:
- PCl3 + 3 ROH → P(OR)3 + 3 HCl
The reaction is general, thus a vast number of such species are known. Phosphites are employed in the Perkow reaction and the Michaelis–Arbuzov reaction. They also serve as ligands in organometallic chemistry.
Intermediate between phosphites and phosphines are phosphonites (P(OR)2R') and phosphinite (P(OR)R'2). Such species arise via alcoholysis reactions of the corresponding phosphinous and phosphonous chlorides ((PClR'2) and PCl2R', respectively).
Phosphines
Main article: PhosphineThe parent compound of the phosphines is PH3, called phosphine in the US and British Commonwealth, but phosphane elsewhere.[5] Replacement of one or more hydrogen centers by an organic substituents (alkyl, aryl), gives PH3−xRx, an organophosphine, generally referred to as phosphines.
-
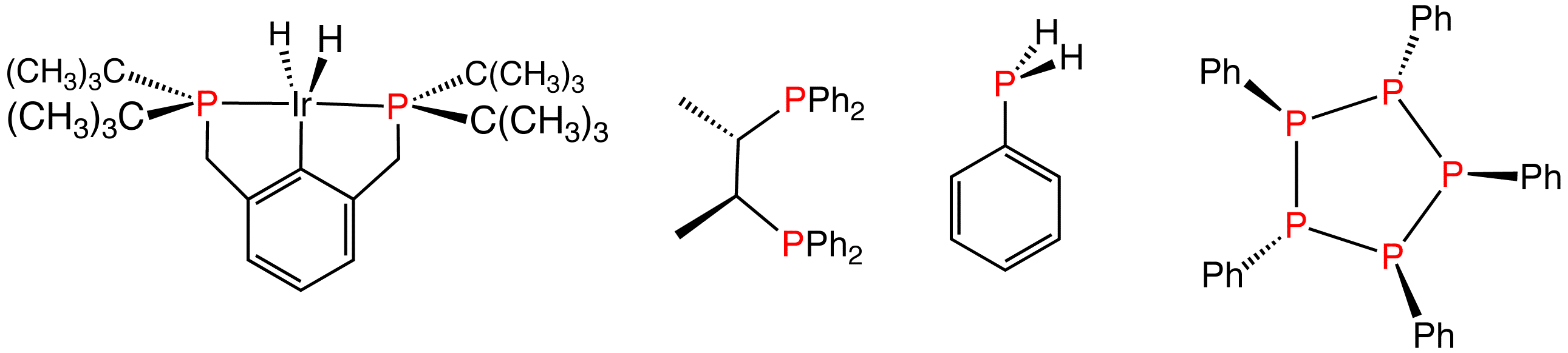
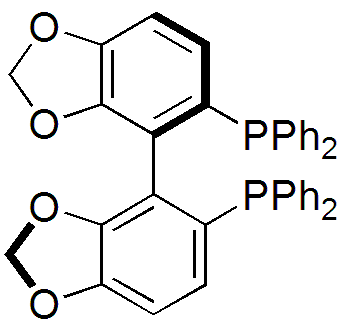
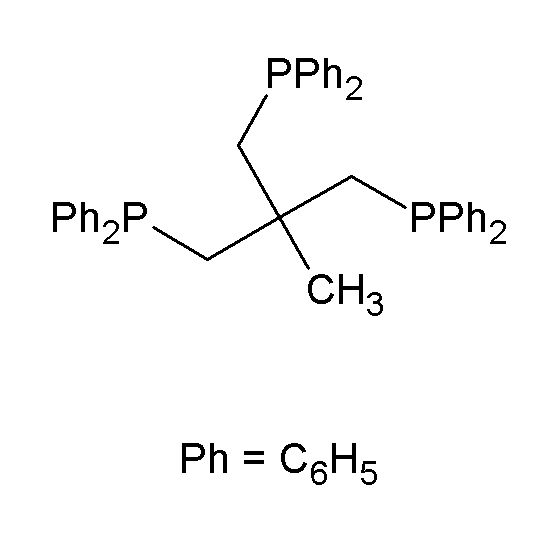
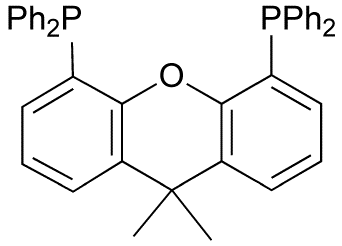
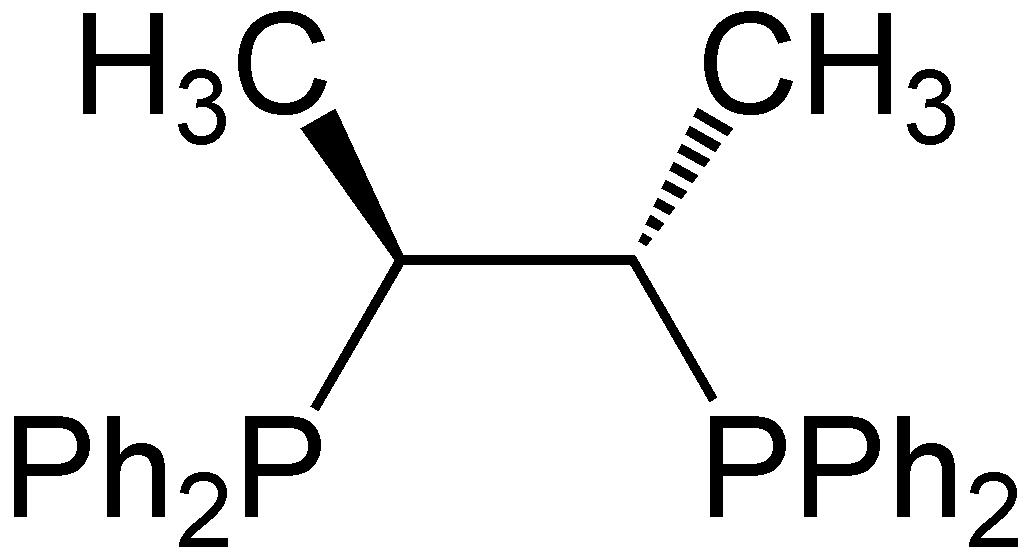
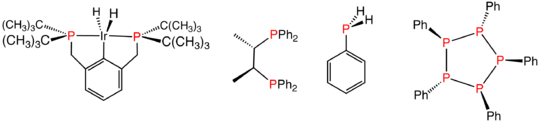 Various reduced organophosphorus compounds: a complex of an organophosphine pincer ligand, the chiral diphosphine used in homogeneous catalysis, the primary phosphine PhPH2, and the phosphorus(I) compound (PPh)5.
Various reduced organophosphorus compounds: a complex of an organophosphine pincer ligand, the chiral diphosphine used in homogeneous catalysis, the primary phosphine PhPH2, and the phosphorus(I) compound (PPh)5.
Comparison of phosphines and amines
The phosphorus atom in phosphines has a formal oxidation state −3 (δ3λ3) and are the phosphorus analogues of amines. Like amines, phosphines have a trigonal pyramidal molecular geometry although often with smaller C-E-C angles (E = N, P), at least in the absence of steric effects. The C-P-C bond angle is 98.6° for trimethylphosphine increasing to 109.7° when the methyl groups are replaced by tert-butyl groups. When used as ligands, the steric bulk of tertiary phosphines is evaluated by their cone angle. The barrier to inversion is also much higher than in amines for a process like nitrogen inversion to occur, and therefore phosphines with three different substituents can be resolved into thermally stable optical isomers. Phosphines are often less basic than corresponding amines, for instance the phosphonium ion itself has a pKa of −14 compared to 9.21 for the ammonium ion; trimethylphosphonium has a pKa of 8.65 compared to 9.76 for trimethylamine. However, triphenylphosphine (pKa 2.73) is more basic than triphenylamine (pKa −5), mainly because the lone pair of the nitrogen in NPh3 is partially delocalized into the three phenyl rings. Whereas the lone pair on nitrogen is delocalized in pyrrole, the lone pair on phosphorus atom in the phosphorus equivalent of pyrrole (phosphole) is not. The reactivity of phosphines matches that of amines with regard to nucleophilicity in the formation of phosphonium salts with the general structure PR4+X−. This property is used in the Appel reaction for converting alcohols to alkyl halides. Phosphines are easily oxidized to the corresponding phosphine oxides, whereas amine oxides are less readily generated. In part for this reason, phosphines are very rarely encountered in nature.
Synthetic routes
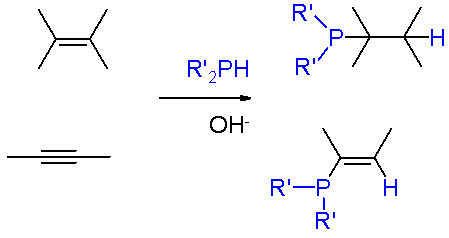
From the commercial perspective, the most important phosphine is triphenylphosphine, several million kilograms being produced annually. It is prepared from the reaction of chlorobenzene, PCl3, and sodium.[4] Phosphines of a more specialized nature are usually prepared by other routes. Phosphorus halides undergo Nucleophilic displacement by organometallic reagents such as Grignard reagents. Conversely, some syntheses entail nucleophilic displacement of phosphide anion equivalents ("R2P-") by aryl- and alkyl halides. Primary (RPH2) and secondary phosphines (RRPH and R2PH) add to alkenes in presence of a strong base (e.g., KOH in DMSO). Markovnikov's rules apply. Similar reactions occur involving alkynes.[6] Base is not required for electron-deficient alkenes (e.g., derivatives of acrylonitrile) and alkynes.
Under free-radical conditions the P-H bonds of primary and secondary phosphines add across alkenes. Such reactions proceed with anti-Markovnikov regiochemistry. AIBN or organic peroxides are used as initiators. Tertiary phosphine oxides and sulfides can be reduced with chlorosilanes and other reagents.
Reactions
The main reaction types of phosphines are as nucleophiles and bases. Their nucleophilicity is evidence by their reactions with alkyl halides to phosphonium salts. Phosphines are nucleophilic catalysts in the dimerization of enones in various reactions in organic synthesis, e.g. the Rauhut–Currier reaction.
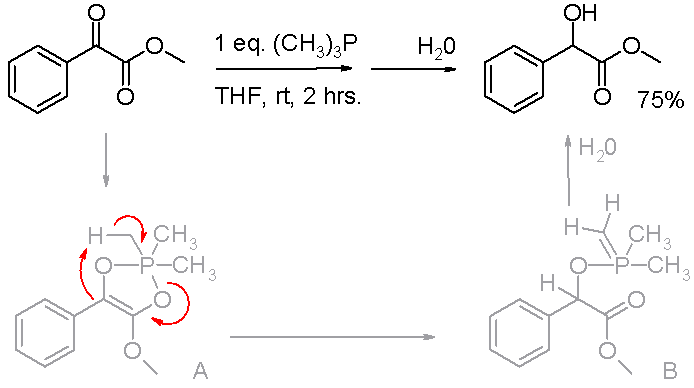
Phosphines are reducing agents, as illustrated in the Staudinger reduction converting azides to amines and in the Mitsunobu reaction for converting alcohols into esters. In these processes, the phosphine is oxidized to the phosphine oxide. Phosphines have also been found to reduce activated carbonyl groups for instance the reduction of an α-keto ester to an α-hydroxy ester in scheme 2.[7] In the proposed reaction mechanism, the first proton is on loan from the methyl group in trimethylphosphine (triphenylphosphine does not react).
-
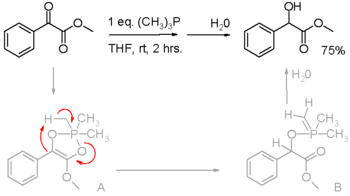 Reduction of activated carbonyl groups by alkyl phosphines
Reduction of activated carbonyl groups by alkyl phosphines
Phosphine ligands
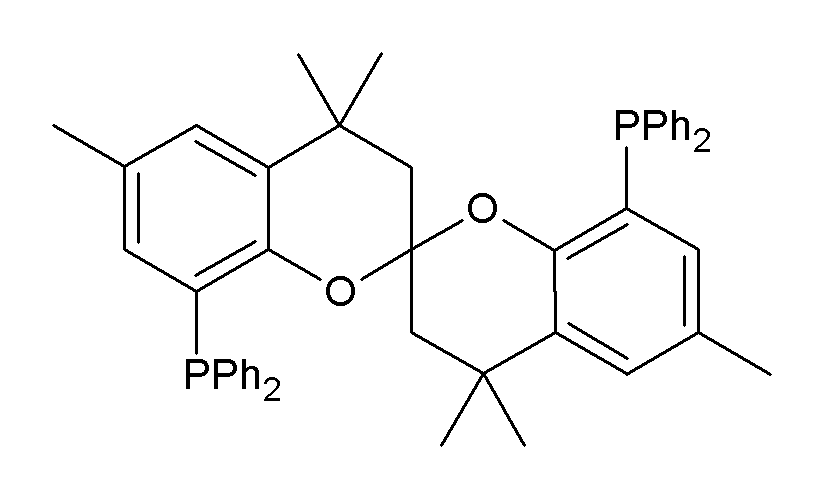
Phosphines such as trimethylphosphine are important ligands in inorganic chemistry. Mainly owing to the utility of asymmetric synthesis, a variety of chiral diphosphines have been popularized, such as BINAP and DIPAMP. A large number of phosphine ligands including diphosphines are simply called "phos ligands".
sPhos 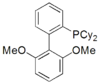
SPANphos 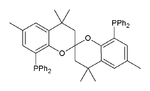
SEGphos 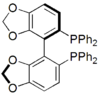
Triphos 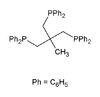
Xantphos 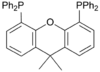
XPhos 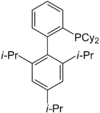
Chiraphos 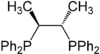
duPhos 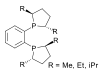
A selection of phos ligands Primary and secondary phosphines
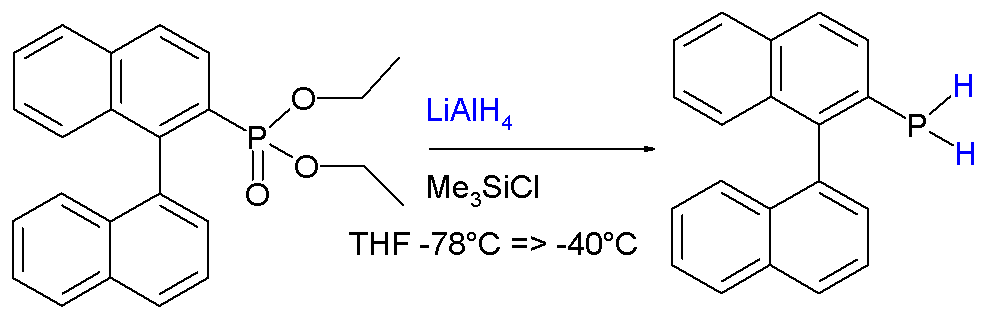
In addition to the other reactions associated with phosphines, those bearing P-H groups exhibit additional reactivity associated with the P-H bonds. They are readily deprotonated using strong bases to give phosphide anions. Primary and secondary phosphines are generally prepared by reduction of related phosphorus halides or esters. For example, phosphonates are reduced to primary phosphines:[8]
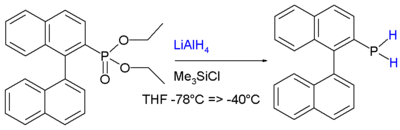 Example of reduction of phosphonate ester to a primary phosphine.
Example of reduction of phosphonate ester to a primary phosphine.The stability is attributed to conjugation between the aromatic ring and the phosphorus lone pair.
Phosphaalkenes and phosphaalkynes
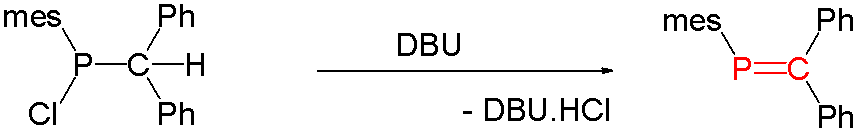
Main articles: Phosphaalkene and PhosphaalkyneCompounds with carbon phosphorus(III) multiple bonds are called phosphaalkenes (R2C=PR) and phosphaalkynes (RC≡P). They are similar in structure, but not in reactivity, to imines (R2C=NR) and nitriles (RC≡N), respectively. In the compound phosphorine, one carbon atom in benzene is replaced by phosphorus. Species of this type are relatively rare but for that reason are of interest to researchers. A general method for the synthesis of phosphaalkenes is by 1,2-elimination of suitable precursors, initiated thermally or by base such as DBU, DABCO, or triethylamine:
Thermolysis of Me2PH generates CH2=PMe, an unstable species in the condensed phase.
Organophosphorus(0), (I), and (II) compounds
Compounds where phosphorus exists in a formal oxidation state of less than III are uncommon, but examples are known for each class. Organophosphorus(0) species are illustrated by the carbene adducts, [P(NHC)]2, where NHC is an N-heterocyclic carbene.[9] With the formulae (RP)n and (R2P)2, respectively, compounds of phosphorus(I) and (II) are generated by reduction of the related organophosphorus(III) chlorides:
Diphosphenes, with the formula R2P2, formally contain phosphorus-phosphorus double bonds. These phosphorus(I) species are rare but are stable provided that the organic substituents are large enough to prevent catenation. Many mixed-valence compounds are known, e.g. the cage P7(CH3)3.
CH He CLi CBe CB CC CN CO CF Ne CNa CMg CAl CSi CP CS CCl CAr CK CCa CSc CTi CV CCr CMn CFe CCo CNi CCu CZn CGa CGe CAs CSe CBr CKr CRb CSr CY CZr CNb CMo CTc CRu CRh CPd CAg CCd CIn CSn CSb CTe CI CXe CCs CBa CHf CTa CW CRe COs CIr CPt CAu CHg CTl CPb CBi CPo CAt Rn Fr Ra Rf Db Sg Bh Hs Mt Ds Rg Cn Uut Uuq Uup Uuh Uus Uuo ↓ CLa CCe CPr CNd CPm CSm CEu CGd CTb CDy CHo CEr CTm CYb CLu Ac Th Pa CU Np Pu Am Cm Bk Cf Es Fm Md No Lr Chemical bonds to carbon Core organic chemistry Many uses in chemistry Academic research, but no widespread use Bond unknown / not assessed See also
- Activity based proteomics a branch of biochemistry that often relies on organophosphorus probes to interrogate enzyme activities
- Organophosphates
- Organothiophosphates
External links
- organophosphorus chemistry @ users.ox.ac.uk; @ www.chem.wisc.edu
- NMR predictor for organophosphorus compound chemical shifts from Alan Brisdon's Research Group Link
References
- ^ Dillon, K. B.; Mathey, F.; Nixon, J. F. Phosphorus. The Carbon Copy; John Wiley & Sons, 1997. ISBN 0-471-97360-2
- ^ Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons, 2000. ISBN 0-471-31824-8
- ^ Racke, K.D., 1992. Degradation of organophosphorus insecticides in environmental matrices. In: Chambers, J.E., Levi, P.E. (Eds.), Organophosphates: Chemistry, Fate, and Effects. Academic Press, San Diego, pp. 47–73.
- ^ a b c Jürgen Svara, Norbert Weferling, Thomas Hofmann “Phosphorus Compounds, Organic” in Ullmann's Encyclopedia of Industrial Chemistry Wiley-VCH, Weinheim, 2006. doi:10.1002/14356007.a19_545.pub2
- ^ Gold Book: Link
- ^ Arbuzova, S. N.; Gusarova, N. K.; Trofimov, B. A. "Nucleophilic and free-radical additions of phosphines and phosphine chalcogenides to alkenes and alkynes." Arkivoc 2006, part v, 12–36 (EL-1761AR). Article
- ^ Zhang, W.; Shi, M. "Reduction of activated carbonyl groups by alkyl phosphines: formation of α-hydroxy esters and ketones." Chem. Commun. 2006, 1218–1220. doi:10.1039/b516467b
- ^ Taming a Functional Group: Creating Air-Stable, Chiral Primary Phosphanes Rachel M. Hiney, Lee J. Higham, Helge Müller-Bunz, Declan G. Gilheany Angewandte Chemie International Edition Volume 45, Issue 43 , Pages 7248 – 7251 2006 doi:10.1002/anie.200602143
- ^ Yuzhong Wang, Yaoming Xie, Pingrong Wei, R. Bruce King, Henry F. Schaefer, III, Paul v. R. Schleyer and Gregory H. Robinson "Carbene-Stabilized Diphosphorus" J. Am. Chem. Soc., 2008, 130, pp 14970–14971. doi:10.1021/ja807828t
P(V)With P-C bonds One P-C bondsP(III)P(V)Two P-C bondsP(III)P(V)Three P-C bondsP(III)P(V)With P-N bonds One P-N bondP(III)P(V)Phosphoramidate (e.g. Phosphocreatine)Two P-N bondP(III)PhosphorodiamiditeP(V)Phosphorodiamidate (e.g. Morpholino, Cyclophosphamide)Three P-N bondP(V)Phosphoramide (e.g. Hexamethylphosphoramide, Metepa)With P-C and P-N bonds One P-C bond and one P-N bondP(V)PhosphonamidateOne P-C bond and two P-N bondP(V)PhosphonamideTwo P-C bond and one P-N bondP(V)PhosphinamideWith P-S bonds One P-S bondP(III)PhosphorothioiteP(V)PhosphorothioateTwo P-S bondP(III)PhosphorodithioiteP(V)PhosphorodithioateCategories: -




Wikimedia Foundation. 2010.