- Organocobalt chemistry
-
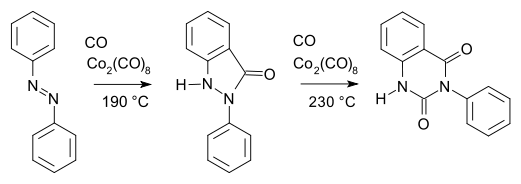
Organocobalt chemistry is the chemistry of organometallic compounds containing a carbon to cobalt chemical bond. Organocobalt compounds are involved in several organic reactions and the important biomolecule vitamin B12 has a cobalt-carbon bond. Many organocobalt compounds are investigated for catalytic properties.[1] An early example of organocobalt chemistry is the carbonylation of azobenzene with dicobalt octacarbonyl as described by Murahashi & Horiie in 1956:[2]
The basic characteristics of organocobalt compounds are:
- ease of forming complexes with alkynes, for example in the compound dicobalt hexacarbonyl diphenylacetylene , ease of forming complexes with diene compounds and with cyclobutadiene compounds, ease formation of sandwich compounds
- high affinity of cobalt for carbonyls. Many cobalt metal carbonyls exist.
- ease of formation of an octahedral molecular geometry with nitrogen or oxygen at the base and Co-C bond formation in the axial position as in vitamin B12.
Contents
Triple bond complexes
Cobalt forms complexes with triple bonded alkynes and cyano compounds. This property is exploited in the use of dicobalt octacarbonyl as protective group for alkynes. In the Nicholas reaction an alkyne group is also protected and at the same time the alpha-carbon position is activated for nucleophilic substitution.
Cyclization reactions
Cobalt compounds react with dialkynes and dienes to cyclic intermediates in cyclometalation. Other alkynes, alkens, nitriles or carbon monoxide can then insert themselves into the Co-C bond. Reaction types based on this concept are the Pauson–Khand reaction (CO insertion) and alkyne trimerization (notably with cyclopentadienylcobalt dicarbonyl).
Carbonylations
Organocobalt compounds are used as catalysts in carbonylation reactions and more specifically in hydroformylation , the formation of aldehydes from an alkene, formaldehyde and hydrogen. An important catalyst in this reaction type is HCo(CO)4 (cobalthydrocarbonyl) at one time used in the industrial production of butyraldehyde from propylene. In these processes cobalt catalysts are competing with rhodium catalysts such as HRh(CO)(PPh3)4]. In hydrocarboxylations hydrogen is replaced by water or an alcohol and the reaction product is a carboxylic acid or an ester. An example of this reaction type is the conversion of butadiene to adipic acid.
Cobalt catalysts (together with iron) are relevant in the Fischer-Tropsch process in which synthesis gas is converted to hydrocarbons. The basic reaction sequence is depicted below [3]:
- M + CO → M-CO (M = Co, Fe)
- M-CO + H2 → M-CH3
- M-CH3 + CO → OC-M-CH3
- OC-M-CH3 → M-(CO)-CH3
- M-(CO)-CH3 + H2 → M-CH2CH3
Vitamin B12-type compounds
In vitamin B12 cobalt has an octahedral geometry with a Co-C bond in an axial position. In methylcobalamin the ligand is a methyl group.
Sandwich compounds
Organocobalt compounds form sandwich compounds. Cobaltocene is a 19-electron metallocene, the compound CoCp(C6(Me)6) ha 20 electrons and 21 electrons are counted in Co(C6(Me)6)2 . The Kläui ligand binds metals.
Cobalt-Mediated Radical Polymerization
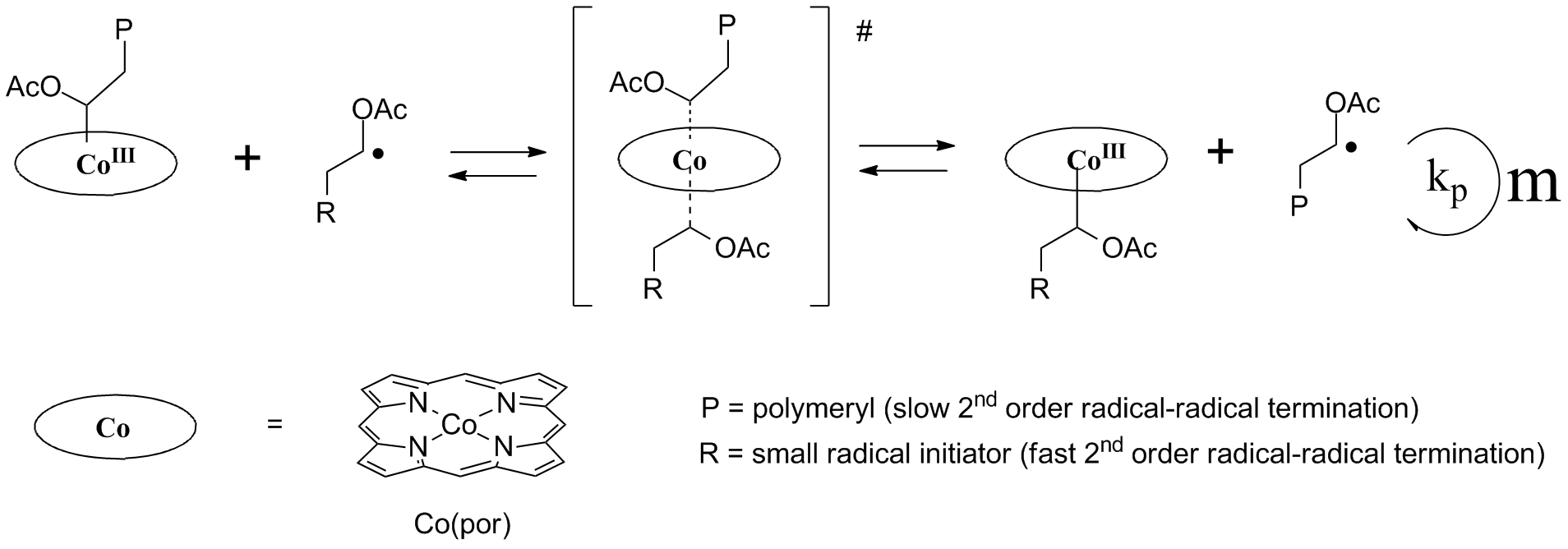
Main article: Cobalt mediated radical polymerizationThe weak cobalt(III)- carbon bond is exploited in so-called cobalt-mediated radical polymerization (CMRP) which is a type of controlled radical polymerization.[4] A Co-C bond containing radical initiator breaks up (by heat or by light) in a carbon free radical and a cobalt(II) radical species. The carbon radical starts polymer chain formation with monomer for instance an alkene as in any ordinary radical polymerization. Cobalt is unusual in that it can reversibly reform a covalent bond with the carbon radical terminus of the growing chain. This reduces the concentration of radicals to a minimum and also undesirable termination reactions by recombination of two carbon radicals. The cobalt trapping reagent is called a persistent radical and the cobalt-capped polymer chain is said to dormant. CMRP can be regarded as a series of carbometalation reactions of vinyl monomers. When the monomer possesses protons that can be easily abstracted by the cobalt radical, (catalytic) chain transfer may occur.
For this reason preferred monomers are acrylic and vinyl esters (e.g. vinyl acetate), acrylic acid and acrylonitrile. The reaction temperature is typically between 0 and 60 °C.
The concept was introduced independently by two groups in 1994.[5][6] Much studied cobalt compounds are cobaloximes, cobalt porphyrins and Co(acac)2 derivatives, used in combination with other radical initiators.
Cobalt mediated radical polymerization can also follow different mechanisms, such as catalytic chain transfer or degenerative transfer:
Cyclotrimerisation
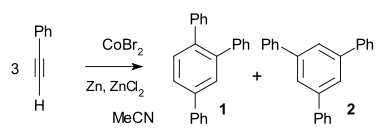
Cobalt compounds are catalysts in alkyne trimerisation. In the example of phenylacetylene a simple catalyst system of cobalt(II) bromide / zinc / zinc iodide suffices to obtain 99% chemical yield and 97% regioselectivity in favor of the ortho substituted reaction product 1.[7]
Mechanism
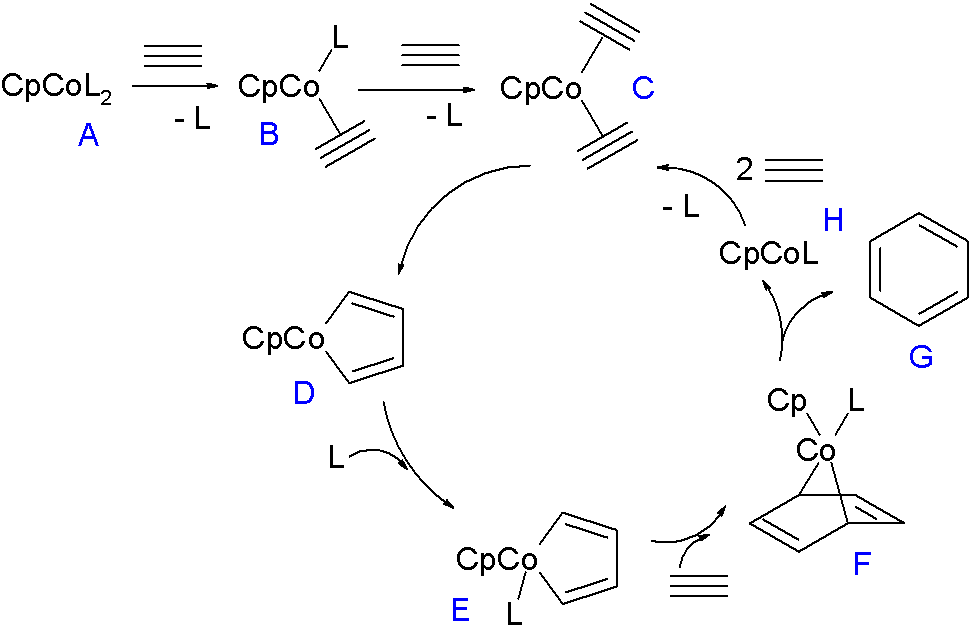
The reaction mechanism for trimerization is fairly well understood. A 2007 mechanism consistent with in silico and experimental data is depicted below for a cobaltocene catalyst [8]
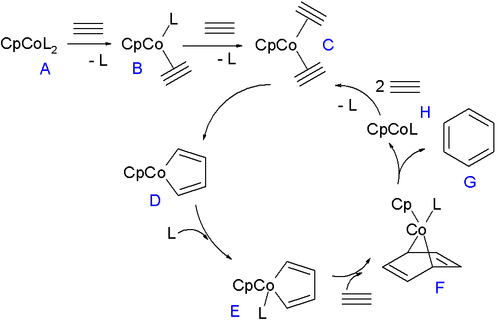
The ligand L in the 18-electron compound CpCoL2 (A) can be triphenylphosphine or carbon monoxide. These ligands are replaced by the alkyne in two steps forming first B and then C which enters the catalytic cycle by oxidative coupling to the 16 VE cobaltacyclopentadiene metallacycle D. This compound forms in its singlet state and therefore first relaxes to the triplet state. This intermediate coordinates to another equivalent of ligand to form E and then forms cobaltanorbornene F (see: norbornadiene) on accepting an alkyne unit in one [4+2]cycloaddition step. In the final step benzene is liberated in a reductive elimination with formation of CpCoL which can re-enter the cycle by accepting two units of alkyne. When the ligand is less of a sigma donor such as ethylene or THF (as a solvent) or when the alkyne is electron-poor as in butynedioic acid a different cycle takes over where another alkyne unit and not a ligand adds to intermediate D. In the remainder of this cycle (not depicted) the new intermediate forms an CpCo(η4arene) complex and then a CpCo(η6arene) sandwich compound before eliminating benzene and CpCo.
Scope
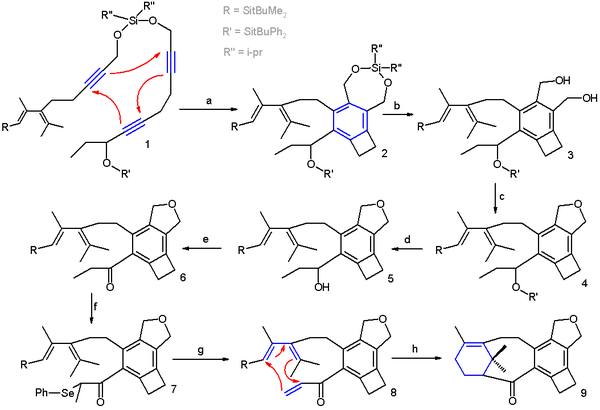
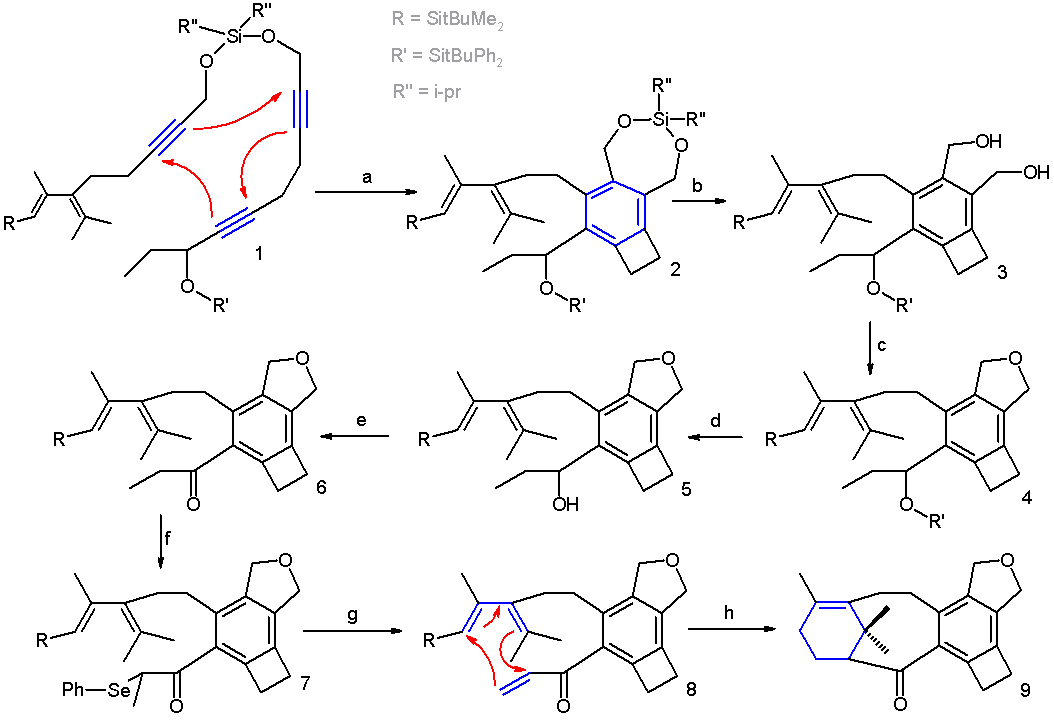
In one study, a combination of a [2+2+2] trimerization and a [4+2] cycloaddition gives access to a Taxol analogue[9] (Scheme 2 [10] ). In this reaction sequence the trimerization takes place in xylene with catalyst the cobaltocene CpCo(CO)2 (one Cp unit replaced by two carbonmonoxide ligands) and with irradiation. The two main components are held together by a temporary silicon tether.
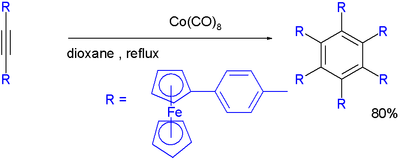
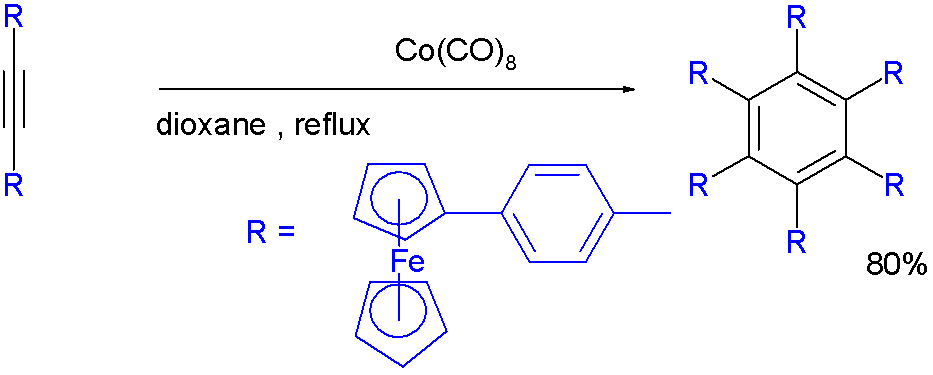
Dicobalt octacarbonyl catalyzes the trimerization towards a hexakis(4-ferrocenylphenyl)benzene [11][12]:
See also
- Chemical bonds of carbon with other elements in the periodic table:
CH He CLi CBe CB CC CN CO CF Ne CNa CMg CAl CSi CP CS CCl CAr CK CCa CSc CTi CV CCr CMn CFe CCo CNi CCu CZn CGa CGe CAs CSe CBr CKr CRb CSr CY CZr CNb CMo CTc CRu CRh CPd CAg CCd CIn CSn CSb CTe CI CXe CCs CBa CHf CTa CW CRe COs CIr CPt CAu CHg CTl CPb CBi CPo CAt Rn Fr Ra Rf Db Sg Bh Hs Mt Ds Rg Cn Uut Uuq Uup Uuh Uus Uuo ↓ CLa CCe CPr CNd CPm CSm CEu CGd CTb CDy CHo CEr CTm CYb CLu Ac Th Pa CU Np Pu Am Cm Bk Cf Es Fm Md No Lr Chemical bonds to carbon Core organic chemistry Many uses in chemistry Academic research, but no widespread use Bond unknown / not assessed References
- ^ Omae, Iwao (2007). "Three characteristic reactions of organocobalt compounds in organic synthesis". Applied Organometallic Chemistry 21 (5): 318. doi:10.1002/aoc.1213.
- ^ Murahashi, Shunsuke; Horiie, Shigeki (1956). Journal of the American Chemical Society 78 (18): 4816. doi:10.1021/ja01599a079.
- ^ Advanced Organic Chemistry F.A. Carey R.J. Sundberg 2nd Ed.
- ^ Antoine, Debuigne; Poli, Rinaldo; Jérôme, Christine; Jérôme, Robert; Detrembleur, Christophe (2009). "Overview of cobalt-mediated radical polymerization: Roots, state of the art and future prospects". Progress in Polymer Science 34 (3): 211–239. doi:10.1016/j.progpolymsci.2008.11.003.
- ^ Wayland, Bradford B.; Poszmik, George; Mukerjee, Shakti L.; Fryd, Michael (1994). "Living Radical Polymerization of Acrylates by Organocobalt Porphyrin Complexes". Journal of the American Chemical Society 116 (17): 7943. doi:10.1021/ja00096a080.
- ^ Arvanitopoulos LD, Greuel MP, Harwood HJ. (1994). ""Living" free radical polymerization using alkyl cobaloximes as photoinitiators". The American Chemical Society 208: 402.
- ^ Gerhard Hilt , Thomas Vogler, Wilfried Hess, Fabrizio Galbiati (2005). "A simple cobalt catalyst system for the efficient and regioselective cyclotrimerisation of alkynes". Chemical Communications 2005 (11): 1474–1475. doi:10.1039/b417832g. PMID 15756340.
- ^ Cobalt-Catalyzed Cyclotrimerization of Alkynes: The Answer to the Puzzle of Parallel Reaction Pathways Nicolas Agenet, Vincent Gandon, K. Peter C. Vollhardt, Max Malacria, and Corinne Aubert J. Am. Chem. Soc.; 2007; 129(28) pp 8860 - 8871; (Article) doi:10.1021/ja072208r
- ^ G. Chouraqui, M. Petit, P. Phansavath, C. Aubert and M. Malacria (2006). "From an Acyclic, Polyunsaturated Precursor to the Polycyclic Taxane Ring System: The [4+2]/[2+2+2] and [2+2+2]/[4+2] Cyclization Strategies". European Journal of Organic Chemistry 2006 (6): 1413–1421. doi:10.1002/ejoc.200500762.
- ^ Reaction sequence: a] CpCo(CO)2, xylene, irradiation b] desilylation by Tetra-n-butylammonium fluoride c] intramolecular ring closure by butyllithium (oxygen nucleophile) and tosyl chloride (tosyl leaving group) e] desilylation by Tetra-n-butylammonium fluoride e] oxidation IBX acid to ketone f] enolate formation with NaHMDS followed by selenation with phenylseleniumchloride g] selenium oxidation and elimination with sodium periodate h] Diels-Alder reaction with boron trifluoride
- ^ V. J. Chebny, D. Dhar, S. V. Lindeman and R. Rathore (2006). "Simultaneous Ejection of Six Electrons at a Constant Potential by Hexakis(4-ferrocenylphenyl)benzene". Org. Lett. 8 (22): 5041–5044. doi:10.1021/ol061904d. PMID 17048838.
- ^ In a redox reaction the six ferrocene substituents lose an electron each at one and the same potential.
Categories:- Organocobalt compounds




Wikimedia Foundation. 2010.