- PDE1
-
PDE1 (phosphodiesterase1) is a phosphodiesterase enzyme also known as calcium- and calmodulin-dependent phosphodiesterase. It is one of the 11 families of phosphodiesterase (PDE1-PDE11). PDE1 has three subtypes, PDE1A, PDE1B and PDE1C which divide further into various isoforms. The various isoforms exhibit different affinities for cAMP and cGMP.[1][2]
Contents
Discovery of PDE1
The existence of the Ca2+-stimulated PDE1 was first demonstrated by Cheung (1970), Kakiuchi and Yamazaki (1970) as a result of their research on bovine brain and rat brain respectively.[1][3]. It has since been found to be widely distributed in various mammalian tissues as well as in other eukaryotes. It is now one of the most intensively studied member of the PDE superfamily of enzymes [3], which today represents 11 gene families [1][4], and the best characterized one as well [3].
Further researches in the field along with increased availability of monoclonal antibodies have shown that various PDE1 isozymes exist and have been identified and purified. It is now known that PDE1 exists as tissue specific isozymes[3].Structure of PDE1
The PDE1 isozyme family belongs to a Class I enzymes [2][5], which includes all vertebrate PDEs and some yeast enzymes.[5] Class I enzymes all have a catalytic core of at least 250 amino acids whereas Class II enzymes lack such a common feature.[5]
Usually vertebrate PDEs are dimers of linear 50 – 150 kDa proteins[5]. They consist of three functional domains; a conserved catalytic core, a regulatory N-terminus and a C-terminus [3-5]. The proteins are chimeric and each domain is associated with their particular function.[2] The regulatory N-terminus is substantially different in various PDE types.[4][5]. They are flanked by the catalytic core and include regions that auto-inhibit the catalytic domains. They also target sequences that control subcellular localization. In PDE1 this region contains a calmodulin binding domain.[4]
The catalytic domains of PDE1 (and other types of PDEs) have three helical subdomains: an N-terminal cyclin-fold region, a linker region and a C-terminal helical bundle. A deep hydrophobic pocket is formed at the interface of these subdomains. It is composed of four subsites. They are: a metal binding site (M site), core pocket (Q pocket), hydrophobic pocket (H pocket) and lid region (L region). The M site is placed at the bottom of the hydrophobic pocket with several metal atoms. The metal atoms bind to residues that are completely conserved in all PDE family members. The identity of the metal atoms is not known with absolute certainty. However, some evidence indicate that at least one of the metals is zinc and the other is likely to be magnesium. The zinc coordination sphere is composed of three histidines, one aspartate and two water molecules. The magnesium coordination sphere involves the same aspartate along with five water molecules, one of which is shared with the zinc molecule. The reputed role of the metal ions include structure stabilization as well as activation of hydroxide to mediate catalysis.[4]
The domains are separated by ”hinge” regions where they can be experimentally separated by limited proteolysis.[2] The PDE1 isozyme family (along with the PDE4 family) is the most diverse one and includes numerous splice variant PDE1 isoforms. It has three subtypes, PDE1A, PDE1B and PDE1C which divide further into various isoforms.[1][2]Localization
The localization of PDE1 isoforms in different tissues/cells and their location within the cells is as follows:
Isoform Tissue/cellular localization Intracellular localization PDE1A (PDE1A) Smooth muscle, heart, lung, brain, sperm [2] Predominantly cytosolic [2] PDE1A1 Heart, lung [2] Predominantly cytosolic [2] PDE1A2 Brain [2] Predominantly cytosolic [2] PDE1B1 (PDE1B) Neurons, lymphocytes, smooth muscle [2] brain, heart, skeletal muscle [1] Cytosolic [2] PDE1B2 Macrophages, lymphocytes [2] Cytosolic [2] PDE1C (PDE1C) Brain, proliferating human smooth muscle, spermatids [2] Cytosolic [2] PDE1C1 Brain, heart, testis [6] - PDE1C2 Olfactory epithelium [2] Cytosolic [2] PDE1C4/5 mRNA is present in the testis [6] - Table 1. Various PDE1s location in tissues and within cells.
Most PDE1 isoforms are reported to be cytosolic. However, there are instances of PDE1s being localized to subcellular regions but little is known about the molecular mechanisms responsible for such localization. It is thought to be likely that the unique N-terminal or C-terminal regions of the various isoforms allow the different proteins to be targeted to specific subcellular domains.[2]
Functional role
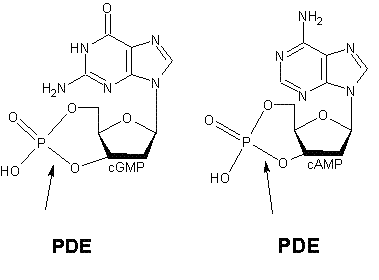
Intracellular second messengers such as cGMP and cAMP undergo rapid changes in concentration in a response to a wide variety of cell specific stimuli. The concentration of these second messengers is determined to a large extent by the relative synthetic activity of adenylate cyclase and degrative activity of cyclic nucleotide PDE.[3] The role of PDE1 enzymes is to degrade both cGMP and cAMP.[7]
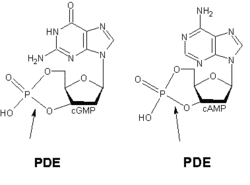 Figure 1: Binding of PDE to cGMP and cAMP respectively
Figure 1: Binding of PDE to cGMP and cAMP respectively
The various isoforms exhibit different affinities for cAMP and cGMP. PDE1A and PDE1B preferentially hydrolyse cGMP, whereas PDE1C degrades both cAMP and cGMP with high affinity. For example in airway smooth muscles of humans and other species, generic PDE1 accounts for more than 50% of the hydrolytic activity of cyclic nucleotides [8]. It has been demonstrated that deletion and overexpression of PDE1 produces strong effects on agonist-induced cAMP signalling but has little effect on the basal cAMP level.[9]
Pharmacology
Because of in vitro regulation by Ca2+/calmodulin, PDE1s are believed to function as a mechanism for integrating cell signalling pathways mediated by cGMP and cAMP with pathways that regulate intracellular calcium levels.[2] The precise function of PDE1 isozymes in various pathophysiological processes is not clear because most of the studies have been carried out in vitro. Therefore it is essential to direct further research to in vivo studies.[3]
PDE1 has been implicated to play a role in a number of physiological and pathological processes:
PDE1A most likely serves to regulate vascular smooth muscle concentration and has been found to be up-regulated in rat aorta in response to chronic nitroglycerin treatment. It is also possible that it plays a role in sperm function.[7]
PDE1B knockout mice have increased locomotor activity and in some paradigms decreased memory and learning abilities. PDE1B is also involved in dopaminergic signalling and is induced in several types of activated immune cells.[7] PDE1B mRNA is induced in PHA or anti-CD3/CD28-activated human T-lymphocytes and participates in IL-13 regulation implicated in allergic diseases.[1]
PDE1C has been shown to be a major regulator of smooth muscle proliferation, at least in human smooth muscle. Nonproliferating smooth muscle cells (SMC) exhibit only low levels of PDE1C expression but it is highly expressed in proliferating SMCs. It can therefore be speculated that inhibition of PDE1C could produce beneficial effects due to its putative inhibition of SMC proliferation, an event that contributes importantly to the pathophysiology of atherosclerosis.[7] Another likely roles of PDE1C is in olfaction [4], to regulate sperm function and neuronal signaling.[7]Regulation
The distinguishing feature of PDE1 as a family is their regulation by calcium (Ca2+) and calmodulin (CaM).[10] Calmodulin has been shown to activate cyclic nucleotide PDE in a calcium-dependent manner and the cooperative binding of four Ca2+ to calmodulin is required to fully activate PDE1 [2]. The binding of one Ca2+/CaM complex per monomer to binding sites near the N-terminus stimulates hydrolysis of cyclic nucleotides. In intact cells, PDE1 is almost exclusively activated by Ca2+ entering the cell from the extracellular space. The regulation of PDE1 by Ca2+ and CaM has been studied in vitro and these studies have shown that eight methionine residues within the hydrophobic clefts of Ca2+-CaM are required for the binding and activation of PDE1. Mutations in the N-terminal lobe of CaM affect its ability to activate PDE1 so it is believed that the C-terminal lobe of CaM serves to target CaM to PDE1, while the N-terminal lobe activates the enzyme. The presence of an aromatic residue, usually a tryptophan, in the CaM-binding region of Ca2+-CaM-regulated proteins may also be required for binding to PDE1.[10]
Between different PDE1 isozymes there is a significant difference in affinity for Ca2+/CaM. In general, the PDE1 enzymes have high affinity for the complex but the affinity can be affected by phosphorylation. Phosphorylation of PDE1A1 and PDE1A2 by protein kinase A and of PDE1B1 by CaM Kinase II decreases their sensitivity to calmodulin activation.[1] This phosphorylation can be reversed by the phosphatase, calcineurin.[2] The phosphorylation of the isozymes is accompanied by a decrease in the isozymes affinity towards CaM, as well as an increase in the Ca2+ concentrations required for CaM activation of the isozymes.[3]PDE1 inhibitors and their function
PDEs have been pursued as therapeutic targets because of the basic pharmacological principle that regulation of degradation of any ligand or second messenger can often make a more rapid and larger percentage change in concentration than comparable rates of synthesis. Another reason is that PDEs do not have to compete with very high levels of endogenous substrate to be effective since the levels of cAMP and cGMP in most cells are typically in the micromolar range.[2]
The availability of high-resolution crystal structures of the catalytic domains of PDEs makes the development of highly potent and specific inhibitors possible.[6]
Many compounds reported as PDE1 inhibitors do not interact directly with the catalytic site of PDE1 but interact during activation, either at the level of calmodulin binding sites such as compound KS505a or directly on Ca2+/calmodulin such as bepril, flunarizine and amiodarone.[1]
Those inhibitors that interact with the catalytic site occupy part of the active site, primarily around the Q pocket and occasionally close to the M pocket.[11] A major point of interaction is a conserved hydrophobic pocket that is involved in orienting the substrate purine ring for interaction with a glutamine residue that is crucial for the catalytic mechanism of the PDEs.[6]
The interactions of inhibitors can be split into three major types: interactions with the metal ions mediated through water, H-bond interactions with the protein residues involved in nucleotide recognition and most importantly the interaction with the hydrophobic residues lining the cavity of the active site. All known inhibitors seem to exploit these three types of interactions and hence these interactions should guide the design of new types of inhibitors.[11]
Initially PDE1 inhibitors were claimed to be effective vascular relaxants. With availability of purified cloned enzymes, however, it is now known that such inhibitors are in fact equally active against PDE5.[4] Those inhibitors include e.g. zaprinast, 8-methoxymethyl IPMX and SCH 51866.[1]
All therapeutically effective PDE inhibitors must be incorporated into the cell because all PDEs are localized in the cytoplasm and/or on intracellular membranes.[7]
Today, there is no real and effective specific PDE1 inhibitor that can be used to assess the functional role of PDE1 in tissues [1].Common PDE1 inhibitors
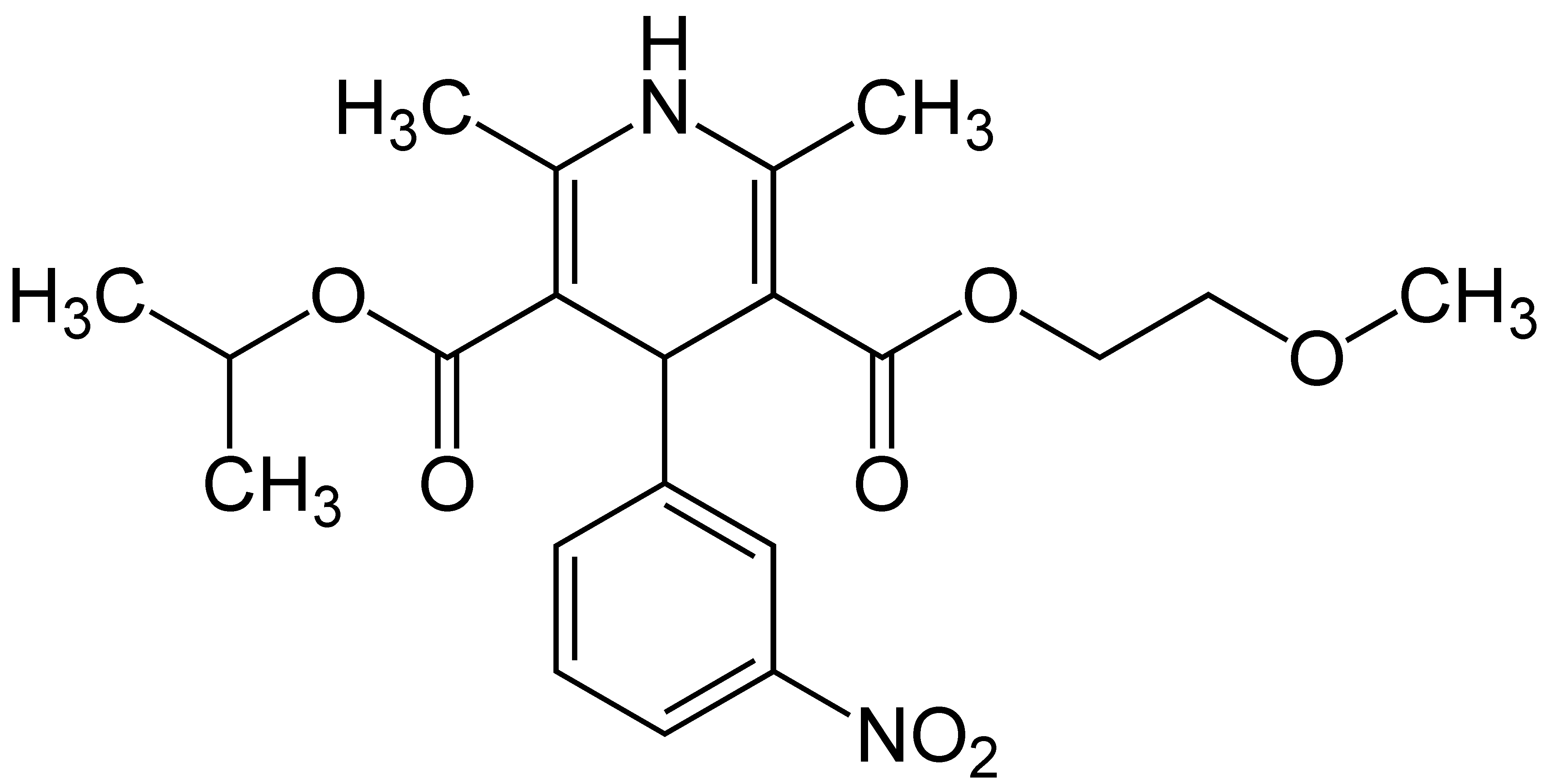
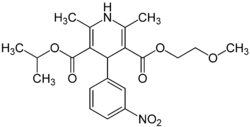 Figure 5. Nimodipine´s structure.
Figure 5. Nimodipine´s structure.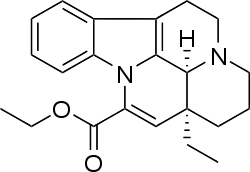 Figure 6. Vinpocetine´s structure.
Figure 6. Vinpocetine´s structure.Nimodipine (3,5-pyridinedicarboxylic acid, 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)-, 2-methoxyethyl 1-methylethyl ester [12]) is a dihydropyridine that antagonizes/blocks specifically L-type Ca2+-channel, and was first described as a PDE1 inhibitor. This effect is not related to its calcium antagonist property since it inhibits, in micromolar range, basal and calmodulin stimulated purified PDE1. Since nimodipine at lower concentrations blocks the L-type calcium channel, it can only be used to estimate PDE1 participation in tissue and cell homogenates [1].
Vinpocetine was described as a specific inhibitor of basal and calmodulin-activated PDE1. This effect leads to an increase of cAMP over cGMP [1][13]. It is mainly used as a pharmacological tool to implicate PDE1. Vinpocetine inhibits differently the various subtypes of PDE1 (IC50 from 8 to 50 µm) and it is also able to inhibit PDE7B. It can not be used as a specific tool to investigate the functional role of PDE1 due to its direct activator effects on BK (Ca) channels.[1] Vinpocetine crosses the blood-brain barrier and is taken up by cerebral tissue. It has been hypothesized that vinpocetine can effect voltage dependent calcium channels.[13]
IC224 inhibits PDE1 (IC50 = 0.08 µM) with a selective ratio of 127 (ratio of IC50 value for the next most sensitive PDE and for IC50 value for PDE1). It was developed by ICOS corporation. If IC224 similarly inhibits basal and calmodulin-activated PDE1 subtypes, this compound could be very helpful to characterize PDE1 activity and to clearly investigate the various roles of PDE1 in pathophysiology.[1]
PDE1 inhibitors in diseases
Nearly all the phosphodiesterases are expressed in the CNS, making this gene family an attractive source of new targets for the treatment of psychiatric and neurodegenerative disorders.[6]
PDE1A2 has a potential role in neurodegenerative diseases e.g.:[4]
Parkinsons
Axonal neurofilament degradation
Motorneuronal degradation
Neuronal ischemia
Alzheimer’s disease
epilepsy
PDE1C could have a role in the regulation of insulin release [5] and may target proliferating smooth muscle cells in atherosclerotic lesions or during restenosis.[4][14]References
- ^ a b c d e f g h i j k l m n Lugnier C (March 2006). "Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents". Pharmacol. Ther. 109 (3): 366–98. doi:10.1016/j.pharmthera.2005.07.003. PMID 16102838. http://linkinghub.elsevier.com/retrieve/pii/S0163-7258(05)00158-0.
- ^ a b c d e f g h i j k l m n o p q r s t u v w Bender AT, Beavo JA (September 2006). "Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use". Pharmacol. Rev. 58 (3): 488–520. doi:10.1124/pr.58.3.5. PMID 16968949. http://pharmrev.aspetjournals.org/cgi/pmidlookup?view=long&pmid=16968949.
- ^ a b c d e f g Kakkar R, Raju RV, Sharma RK (July 1999). "Calmodulin-dependent cyclic nucleotide phosphodiesterase (PDE1)". Cell. Mol. Life Sci. 55 (8-9): 1164–86. doi:10.1007/s000180050364. PMID 10442095. http://link.springer.de/link/service/journals/00018/bibs/90558-9/90551164.htm.
- ^ a b c d e f g Jeon YH, Heo YS, Kim CM, et al. (June 2005). "Phosphodiesterase: overview of protein structures, potential therapeutic applications and recent progress in drug development". Cell. Mol. Life Sci. 62 (11): 1198–220. doi:10.1007/s00018-005-4533-5. PMID 15798894.
- ^ a b c d e f Dousa TP (January 1999). "Cyclic-3',5'-nucleotide phosphodiesterase isozymes in cell biology and pathophysiology of the kidney". Kidney Int. 55 (1): 29–62. doi:10.1046/j.1523-1755.1999.00233.x. PMID 9893113.
- ^ a b c d e Menniti FS, Faraci WS, Schmidt CJ (August 2006). "Phosphodiesterases in the CNS: targets for drug development". Nat Rev Drug Discov 5 (8): 660–70. doi:10.1038/nrd2058. PMID 16883304.
- ^ a b c d e f Bischoff E (June 2004). "Potency, selectivity, and consequences of nonselectivity of PDE inhibition". Int. J. Impot. Res. 16 (Suppl 1): S11–4. doi:10.1038/sj.ijir.3901208. PMID 15224129.
- ^ Giembycz MA (June 2005). "Life after PDE4: overcoming adverse events with dual-specificity phosphodiesterase inhibitors". Curr Opin Pharmacol 5 (3): 238–44. doi:10.1016/j.coph.2005.04.001. PMID 15907909. http://linkinghub.elsevier.com/retrieve/pii/S1471-4892(05)00037-8.
- ^ Thevelein JM, de Winde JH (September 1999). "Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae". Mol. Microbiol. 33 (5): 904–18. doi:10.1046/j.1365-2958.1999.01538.x. PMID 10476026. http://www3.interscience.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0950-382X&date=1999&volume=33&issue=5&spage=904.
- ^ a b Goraya TA, Cooper DM (July 2005). "Ca2+-calmodulin-dependent phosphodiesterase (PDE1): current perspectives". Cell. Signal. 17 (7): 789–97. doi:10.1016/j.cellsig.2004.12.017. PMID 15763421. http://linkinghub.elsevier.com/retrieve/pii/S0898-6568(04)00293-1.
- ^ a b Card GL, England BP, Suzuki Y, et al. (December 2004). "Structural basis for the activity of drugs that inhibit phosphodiesterases". Structure 12 (12): 2233–47. doi:10.1016/j.str.2004.10.004. PMID 15576036. http://linkinghub.elsevier.com/retrieve/pii/S0969212604003727.
- ^ On-line Medical Dictionary
- ^ a b "Vinpocetine. Monograph". Altern Med Rev 7 (3): 240–3. June 2002. PMID 12126465.
- ^ Matsumoto T, Kobayashi T, Kamata K (August 2003). "Phosphodiesterases in the vascular system". J Smooth Muscle Res 39 (4): 67–86. doi:10.1540/jsmr.39.67. PMID 14692693. http://joi.jlc.jst.go.jp/JST.JSTAGE/jsmr/39.67?lang=en&from=PubMed.
Categories:- EC 3.1.4
- Molecular biology

Wikimedia Foundation. 2010.