- Congenital adrenal hyperplasia
-
CAH redirects here. For other meanings of CAH see CAH (disambiguation)
Congenital adrenal hyperplasia Classification and external resources ICD-10 E25.0 ICD-9 255.2 OMIM 201910 201710 202110 201810 202010 DiseasesDB 1854 1832 4 1841 2565 MedlinePlus 000411 eMedicine ped/48 MeSH D000312 Congenital adrenal hyperplasia (CAH) refers to any of several autosomal recessive diseases resulting from mutations of genes for enzymes mediating the biochemical steps of production of cortisol from cholesterol by the adrenal glands (steroidogenesis).[1] CAH is one of the possible underlying synthesis problems in Addison’s disease. CAH is a genetic disorder in which girls are masculinized because the adrenal glands secrete large amounts of androgen during prenatal development. The extra androgen does not affect a baby boy's physical development, but in baby girls it can enlarge the clitoris so that it resembles a penis. The girls sometimes have surgery during infancy to correct their physical appearance, although this practice is highly controversial, and they can receive hormone therapy to correct the imbalance of androgen. During childhood and adolescence, girls with CAH prefer masculine activities and male playmates to a much greater extent than girls not exposed to these amounts of androgen. [2]
Most of these conditions involve excessive or deficient production of sex steroids and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults.[3]
Contents
Associated conditions
The symptoms of CAH vary depending upon the form of CAH and the gender of the patient. Symptoms can include:
Due to inadequate mineralocorticoids:
- vomiting due to salt-wasting leading to dehydration and death
Due to excess androgens:- ambiguous genitalia, in some females, such that it can be initially difficult to determine sex
- early pubic hair and rapid growth in childhood
- precocious puberty or failure of puberty to occur (sexual infantilism: absent or delayed puberty)
- excessive facial hair, virilization, and/or menstrual irregularity in adolescence
- infertility due to anovulation
- enlarged clitoris and shallow vagina[4]
Classification
Cortisol is an adrenal steroid hormone that is required for normal endocrine function. Production begins in the second month of fetal life. Poor cortisol production is a hallmark of most forms of CAH. Inefficient cortisol production results in rising levels of ACTH, which in turn induces overgrowth (hyperplasia) and overactivity of the steroid-producing cells of the adrenal cortex. The defects causing adrenal hyperplasia are congenital (i.e., present at birth).
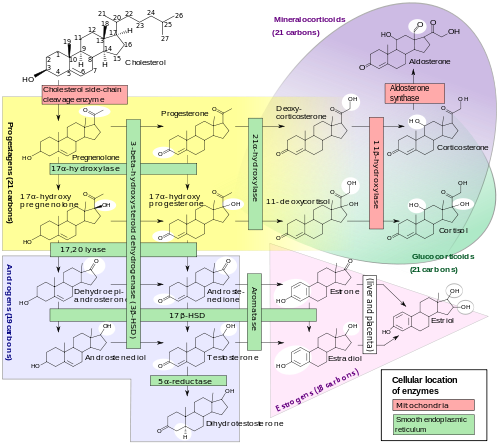 Steroidogenesis. The enzymes affected in CAH are represented by one red and four green bars on the top half of the diagram (for example, "21α-hydroxylase" is visible near the top center. "17α-hydroxylase" and "17,20 lyase" are carried out by a single enzyme.) Depending upon which enzyme is unavailable, there is a reduced production of androgens (lower left) or mineralocorticoids (upper right). This in turn can lead to increased production of other molecules, due to a buildup of precursors.
Steroidogenesis. The enzymes affected in CAH are represented by one red and four green bars on the top half of the diagram (for example, "21α-hydroxylase" is visible near the top center. "17α-hydroxylase" and "17,20 lyase" are carried out by a single enzyme.) Depending upon which enzyme is unavailable, there is a reduced production of androgens (lower left) or mineralocorticoids (upper right). This in turn can lead to increased production of other molecules, due to a buildup of precursors.
Cortisol deficiency in CAH is usually partial, and not the most serious problem for an affected person. Synthesis of cortisol shares steps with synthesis of mineralocorticoids such as aldosterone, androgens such as testosterone, and estrogens such as estradiol. The resulting excessive or deficient production of these three classes of hormones produce the most important problems for people with CAH. Specific enzyme inefficiencies are associated with characteristic patterns of over- or underproduction of mineralocorticoids or sex steroids.
Common medical term % OMIM Enzyme(s) Locus Substrate(s) Product(s) Mineralocorticoids Androgens 21-hydroxylase CAH 90-95% 201910 P450c21 6p21.3 17OH-progesterone→
progesterone→11-deoxycortisol
DOC↓ ↑ 11β-hydroxylase CAH 5% 202010 P450c11β 8q21-22 11-deoxycortisol→
DOC→cortisol
corticosterone↑ ↑ 3β-HSD CAH very rare 201810 3βHSD II 1p13 pregnenolone→
17OH-pregnenolone→
DHEA→progesterone
17OH-progesterone
androstenedione↓ ↓ 17α-hydroxylase CAH very rare 202110 P450c17 10q24.3 pregnenolone→
progesterone→
17OH-pregnenolone→17OH-pregnenolone
17OH-progesterone
DHEA↑ ↓ lipoid CAH
(20,22-desmolase)very rare 201710 StAR
P450scc8p11.2
15q23-q24transport of cholesterol
cholesterol→into mitochondria
pregnenolone↓ ↓ Since the 1960s most endocrinologists have referred to the forms of CAH by the traditional names in the left column, which generally correspond to the deficient enzyme activity. As exact structures and genes for the enzymes were identified in the 1980s, most of the enzymes were found to be cytochrome P450 oxidases and were renamed to reflect this. In some cases, more than one enzyme was found to participate in a reaction, and in other cases a single enzyme mediated in more than one reaction. There was also variation in different tissues and mammalian species.
In all its forms, congenital adrenal hyperplasia due to 21-hydroxylase deficiency accounts for about 95% of diagnosed cases of CAH. Unless another specific enzyme is mentioned, "CAH" in nearly all contexts refers to 21-hydroxylase deficiency. (The terms "salt-wasting CAH", and "simple virilizing CAH" usually refer to subtypes of this condition.) CAH due to deficiencies of enzymes other than 21-hydroxylase present many of the same management challenges as 21-hydroxylase deficiency, but some involve mineralocorticoid excess or sex steroid deficiency.
Penetrance
Further variability is introduced by the degree of enzyme inefficiency produced by the specific alleles each patient has. Some alleles result in more severe degrees of enzyme inefficiency. In general, severe degrees of inefficiency produce changes in the fetus and problems in prenatal or perinatal life. Milder degrees of inefficiency are usually associated with excessive or deficient sex hormone effects in childhood or adolescence, while the mildest form of CAH interferes with ovulation and fertility in adults.
Treatment
Treatment of all forms of CAH may include any of:
- supplying enough glucocorticoid to reduce hyperplasia and overproduction of androgens or mineralocorticoids
- providing replacement mineralocorticoid and extra salt if the person is deficient
- providing replacement testosterone or estrogen at puberty if the person is deficient
- additional treatments to optimize growth by delaying puberty or delaying bone maturation
All of these management issues are discussed in more detail in congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
Dexamethasone is used as an off-label early pre-natal treatment for the symptoms of CAH in female fetuses, but it does not treat the underlying congenital disorder. A 2007 Swedish clinical trial found that treatment may cause cognitive and behavioural defects, but the small number of test subjects means the study cannot be considered definitive. Administration of pre-natal dexamethasone has been the subject of controversy over issues of informed consent and because treatment must predate a clinical diagnosis of CAH in the female fetus.[5] The treatment has also raised concerns in the LGBT community following an essay posted to the forum of the Hastings Center, a think tank devoted to bioethics, which quoted published research that suggested that pre-natal treatment of female fetuses could prevent those fetuses from becoming lesbians after birth, may make them more likely to engage in "traditionally" female-identified behaviour and careers, and more interested in bearing and raising children. Citing a known attempt by a man using his knowledge of the fraternal birth order effect to avoid having a homosexual son by using a surrogate, the essayists (Professor Alice Dreger of Northwestern University's Feinberg School of Medicine, Professor Ellen Feder of American University and attorney Anne Tamar-Mattis) suggest that pre-natal "dex" treatments constitute the first known attempt to use in utero protocols to reduce the incidence of homosexuality and bisexuality in humans.[6] They find such tampering to be morally objectionable. Others[who?] feel this is a proven valid way of preventing homosexuality.
Since CAH is a recessive gene, both the mother and father must be recessive carriers of CAH for a child to have CAH. Due to advances in modern medicine, those couples with the recessive CAH genes have an option to prevent CAH in their offspring through preimplantation genetic diagnosis (PGD). In PGD, the egg is fertilized outside the women's body in a petri dish (IVF). On the 3rd day, when the embryo has developed from one cell to about 4 to 6 cells, one of those cells is removed from the embryo without harming the embryo. The embryo continues to grow until day 5 when it is either frozen or implanted into the mother. Meanwhile, the removed cell is analyzed to determine if the embryo has CAH. If the embryo is determined to have CAH, the parents may make a decision as to whether they wish to have it implanted in the mother or not.[citation needed]
Screening for CAH in neonates
Currently, in the United States and over 40 other countries, every child born is screened for CAH at birth. This test will detect elevated levels of 17-hydroxy-progesterone (17-OHP). Detecting high levels of 17-OHP enables early detection of CAH. Newborns detected early enough can be placed on medication and live a relatively normal life.
History
Before 20th century
An Italian anatomist, Luigi De Crecchio provided the earliest known description of a case of probable CAH.
I propose in this narrative that it is sometimes extremely difficult and even impossible to determine sex during life. In one of the anatomical theaters of the hospital..., there arrived toward the end of January a cadaver which in life was the body of a certain Joseph Marzo... The general physiognomy was decidedly male in all respects. There were no feminine curves to the body. There was a heavy beard. There was some delicacy of structure with muscles that were not very well developed... The distribution of pubic hair was typical of the male. Perhaps the lower extremities were somewhat delicate, resembling the female, and were covered with hair... The penis was curved posteriorly and measured 6 cm, or with stretching, 10 cm. The corona was 3 cm long and 8 cm in circumference. There was an ample prepuce. There was a first grade hypospadias... There were two folds of skin coming from the top of the penis and encircling it on either side. These were somewhat loose and resembled labia majora.
De Crecchio then described the internal organs, which included a normal vagina, uterus, tubes, and ovaries.
It was of the greatest importance to determine the habits, tendencies, passions, and general character of this individual... I was determined to get as complete a story as possible, determined to get at the base of the facts and to avoid undue exaggeration which was rampant in the conversation of many of the people present at the time of the dissection.
He interviewed many people and satisfied himself that Joseph Marzo "conducted himself within the sexual area exclusively as a male, "even to the point of contracting the "French disease" on two occasions. The cause of death was another in a series of episodes of vomiting and diarrhea.[7]
This account was translated by Alfred Bongiovanni from De Crecchio (Sopra un caso di apparenzi virili in una donna. Morgagni 7:154-188, 1865) in 1963 for an article in the New England Journal of Medicine.
20th and 21st century
The association of excessive sex steroid effects with diseases of the adrenal cortex have been recognized for over a century. The term adrenogenital syndrome was applied to both sex-steroid producing tumors and severe forms of CAH for much of the 20th century, before some of the forms of CAH were understood. Congenital adrenal hyperplasia, which also dates to the first half of the century, has become the preferred term to reduce ambiguity and to emphasize the underlying pathophysiology of the disorders.
Much of our modern understanding and treatment of CAH comes from research conducted at Johns Hopkins Medical School in Baltimore in the middle of the 20th century. Lawson Wilkins, "founder" of pediatric endocrinology, worked out the apparently paradoxical pathophysiology: that hyperplasia and overproduction of adrenal androgens resulted from impaired capacity for making cortisol. He reported use of adrenal cortical extracts to treat children with CAH in 1950. Genital reconstructive surgery was also pioneered at Hopkins. After application of karyotyping to CAH and other intersex disorders in the 1950s, John Money, JL Hampson, and JG Hampson persuaded both the scientific community and the public that sex assignment should not be based on any single biological criterion, and gender identity was largely learned and has no simple relationship with chromosomes or hormones. See Intersex for a fuller history, including recent controversies over reconstructive surgery.
Hydrocortisone, fludrocortisone, and prednisone were available by the late 1950s. By 1980 all of the relevant steroids could be measured in blood by reference laboratories for patient care. By 1990 nearly all specific genes and enzymes had been identified.
However, the last decade has seen a number of new developments, discussed more extensively in congenital adrenal hyperplasia due to 21-hydroxylase deficiency:
- debate over the value of genital reconstructive surgery and changing standards
- debate over sex assignment of severely virilized XX infants
- new treatments to improve height outcomes
- newborn screening programs to detect CAH at birth
- increasing attempts to treat CAH before birth
References
- ^ David A. Warrell (2005). Oxford textbook of medicine: Sections 18-33. Oxford University Press. pp. 261–. ISBN 9780198569787. http://books.google.com/books?id=hL1NKQJlY1IC&pg=PA261. Retrieved 14 June 2010.
- ^ Kail, R.V., & Cavanaugh, J.C. (2007). Human Development: A Life-Span View (5th ed.). Belmont, CA: Wadsworth, Cengage Learning.
- ^ Aubrey Milunsky; Jeff Milunsky (29 January 2010). Genetic Disorders and the Fetus: Diagnosis, Prevention and Treatment. John Wiley and Sons. pp. 600–. ISBN 9781405190879. http://books.google.com/books?id=oKCmA4dOYtMC&pg=PA600. Retrieved 14 June 2010.
- ^ Richard D. McAnulty, M. Michele Burnette (2006) Sex and sexuality, Volume 1, Greenwood Publishing Group, p.165
- ^ Elton, Catherine (2010-06-18). "A Prenatal Treatment Raises Questions of Medical Ethics". TIME. http://www.time.com/time/health/article/0,8599,1996453,00.html?xid=rss-mostpopular. Retrieved 2010-07-05.
- ^ Dreger, Alice; Ellen K. Feder, Anne Tamar-Mattis (2010-06-29). "Preventing Homosexuality (and Uppity Women) in the Womb?". Bioethics Forum, a service of the Hastings Center. http://www.thehastingscenter.org/Bioethicsforum/Post.aspx?id=4754&blogid=140#ixzz0sqEoOR95. Retrieved 2010-07-05.
- ^ Bongiovanni, Alfred M.; Root, Allen W. (1963). "The Adrenogenital Syndrome". New England Journal of Medicine 268 (23): 1283. doi:10.1056/NEJM196306062682308.
External links
Inborn error of steroid metabolism Mevalonate pathway To cholesterol 7-Dehydrocholesterol path: Hydrops-ectopic calcification-moth-eaten skeletal dysplasia · CHILD syndrome · Conradi-Hünermann syndrome · Lathosterolosis · Smith-Lemli-Opitz syndrome
desmosterol path: DesmosterolosisSteroids Corticosteroid
(including CAH)aldosterone: Glucocorticoid remediable aldosteronism
cortisol/cortisone: CAH 17α hydroxylase · CAH 11β hydroxylase
both: CAH 3β dehydrogenase · CAH 21α hydroxylase · Apparent mineralocorticoid excess syndrome/11β dehydrogenaseTo androgensTo estrogensAromatase deficiencyOtherCategories:- Pediatrics
- Adrenal gland disorders
- Endocrine-related cutaneous conditions
- Autosomal recessive disorders
- Intersexuality

Wikimedia Foundation. 2010.