- Glycogen phosphorylase
-
Phosphorylase 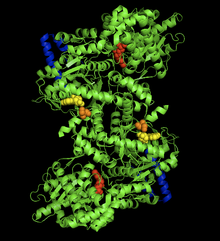
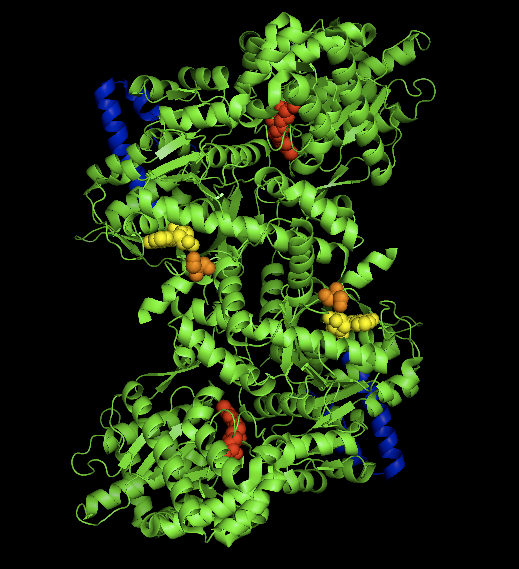
The crystal structure of the rabit muscle glycogen phosphorylase-AMP complex. AMP allosteric site (yellow), phosporylated Ser14 (orange), glycogen binding site (blue), catalytic site (red).[1] Identifiers EC number 2.4.1.1 CAS number 9035-74-9 Databases IntEnz IntEnz view BRENDA BRENDA entry ExPASy NiceZyme view KEGG KEGG entry MetaCyc metabolic pathway PRIAM profile PDB structures RCSB PDB PDBe PDBsum Gene Ontology AmiGO / EGO Search PMC articles PubMed articles Glycogen phosphorylase is one of the phosphorylase enzymes (EC 2.4.1.1). Glycogen phosphorylase catalyzes the rate-limiting step in the degradation of glycogen in animals by releasing glucose-1-phosphate from the terminal alpha-1,4-glycosidic bond. Glycogen phosphorylase is also studied as a model protein regulated by both reversible phosphorylation and allosteric effects.
Contents
Mechanism
The overall reaction is written as:
(α-1,4 glycogen chain)n + Pi ↔ (α-1,4 glycogen chain)n-1 + D-glucose-1-phosphate.[2]
Glycogen phosphorylase breaks up glycogen into glucose subunits. Glycogen is left with one fewer glucose molecule, and the free glucose molecule is in the form of glucose-1-phosphate. In order to be used for metabolism, it must be converted to glucose-6-phosphate by the enzyme phosphoglucomutase.
Although the reaction is reversible in solution, within the cell the enzyme only works in the forward direction as shown above because the concentration of inorganic phosphate is much higher than that of glucose-1-phosphate.[2]

Glycogen phosphorylase can act only on linear chains of glycogen (α1-4 glycosidic linkage). Its work will immediately come to a halt four residues away from α1-6 branch (which are exceedingly common in glycogen). In these situations, a debranching enzyme is necessary, which will straighten out the chain in that area. In addition, the enzyme transferase shifts a block of 3 glucosyl residues from the outer branch to the other end, and then a α1-6 glucosidase enzyme is required to break the remaining (single glucose) α1-6 residue that remains in the new linear chain. After all this is done, glycogen phosphorylase can continue. The enzyme is specific to α1-4 chains, as the molecule contains a 30-angstrom-long crevice with the same radius as the helix formed by the glycogen chain; this accommodates 4-5 glucosyl residues, but is too narrow for branches. This crevice connects the glycogen storage site to the active, catalytic site.
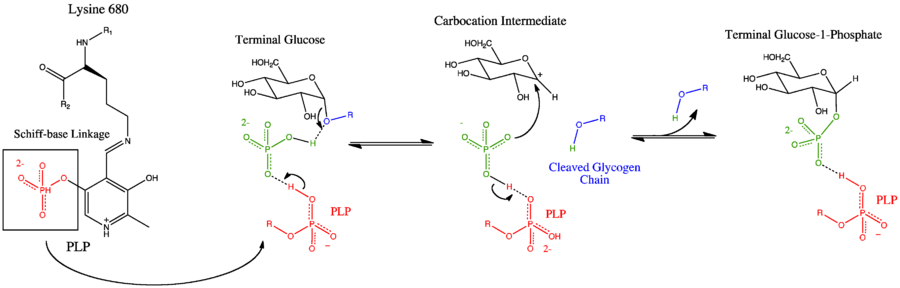
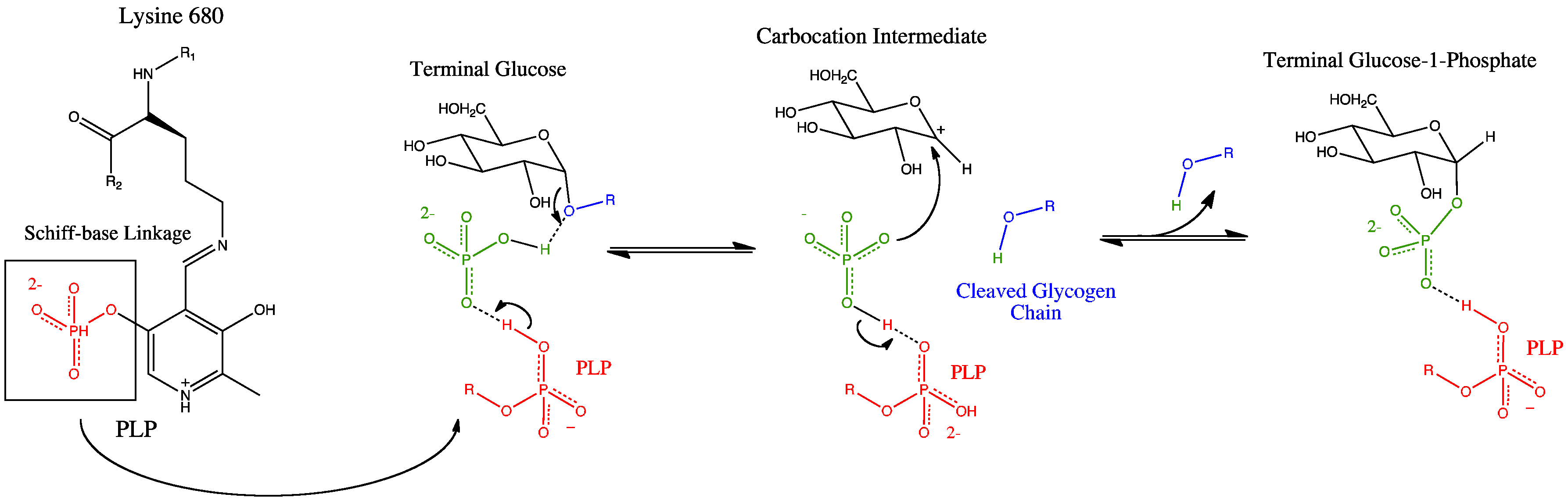
Glycogen phosphorylase has a pyridoxal phosphate (PLP, derived from Vitamin B6) at each catalytic site. Pyridoxal phosphate links with basic residues (in this case Lys680) and covalently forms a Schiff base. Once the Schiff base linkage is formed, holding the PLP molecule in the active site, the phosphate group on the PLP readily donates a proton to an inorganic phosphate molecule, allowing the inorganic phosphate to in turn be deprotonated by the oxygen forming the α-1,4 glycosidic linkage. PLP is readily deprotonated because its negative charge is not only stabilized within the phosphate group, but also in the pyridine ring, thus the conjugate base resulting from the deprotonation of PLP is quite stable. The protonated oxygen now represents a good leaving group, and the glycogen chain is separated from the terminal glycogen in an SN1 fashion, resulting in the formation of a glucose molecule with a secondary carbocation at the 1 position. Finally, the deprotonated inorganic phosphate acts as a nucleophile and bonds with the carbocation, resulting in the formation of glucose-1-phosphate and a glycogen chain shortened by one glucose molecule.
There is also an alternative proposed mechanism involving a positively charged oxygen in a half-chair conformation. [3]
Structure
The glycogen phosphorylase monomer is a large protein, composed of 842 amino acids with a mass of 97.434 kDa in muscle cells. While the enzyme can exist as an inactive monomer or tetramer, it is biologically active as a dimer of two identical subunits.[4]
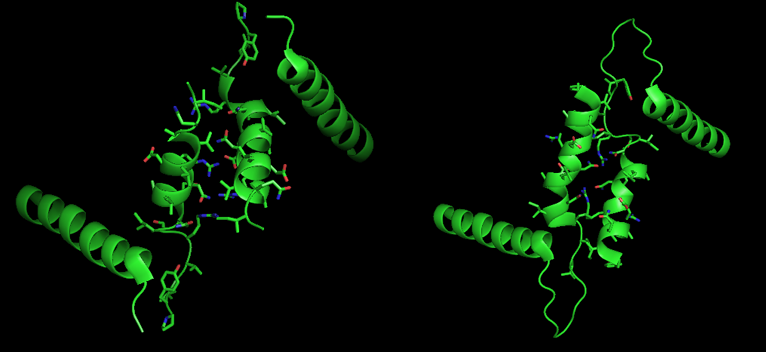
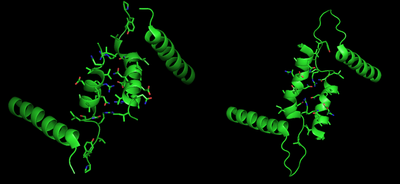 R and T States of Glycogen Phosphorylase b Tower Helices, on the left and right respectively. Note the relative positioning of the central tower helices, as well as the increased interactions between subunits in the R state. PDB3CEH, PDB3E3O
R and T States of Glycogen Phosphorylase b Tower Helices, on the left and right respectively. Note the relative positioning of the central tower helices, as well as the increased interactions between subunits in the R state. PDB3CEH, PDB3E3O
The glycogen phosphorylase dimer has many regions of biological significance, including catalytic sites, glycogen binding sites, allosteric sites, and a reversibly phosphorylated serine residue. First, the catalytic sites are relatively buried, 15Å from the surface of the protein and from the subunit interface.[5] This lack of easy access of the catalytic site to the surface is significant in that it makes the protein activity highly susceptible to regulation, as small allosteric effects could greatly increase the relative access of glycogen to the site.
Perhaps the most important regulatory site is Ser14, the site of reversible phosphorylation very close to the subunit interface. The structural change associated with phosphorylation, and with the conversion of phosphorylase b to phosphorylase a, is the arrangement of the originally disordered residues 10 to 22 into α helices. This change increases phosphorylase activity up to 25% even in the absence of AMP, and enhances AMP activation further.[6]
The allosteric site of AMP binding on muscle isoforms of glycogen phosphorylase are close to the subunit interface just like Ser14. Binding of AMP at this site, corresponding in a change from the T state of the enzyme to the R state, results in small changes in tertiary structure at the subunit interface leading to large changes in quaternary structure.[7] AMP binding rotates the tower helices (residues 262-278) of the two subunits 50˚ relative to one another through greater organization and intersubunit interactions. This rotation of the tower helices leads to a rotation of the two subunits by 10˚ relative to one another, and more importantly disorders residues 282-286 (the 280s loop) that block access to the catalytic site in the T state but do not in the R state.[5]
The final, perhaps most curious site on the glycogen phosphorylase protein is the so-called glycogen storage site. Residues 397-437 form this structure, which allows the protein to covalently bind to the glycogen chain a full 30 Å from the catalytic site . This site is most likely the site at which the enzyme binds to glycogen granules before initiating cleavage of terminal glucose molecules. In fact, 70% of dimeric phosphorylase in the cell exists as bound to glycogen granules rather than free floating.[8]
In mammals, the major isozymes of glycogen phosphorylase are found in muscle, liver, and brain. The brain type is predominant in adult brain and embryonic tissues, whereas the liver and muscle types are predominant in adult liver and skeletal muscle, respectively.[9]
Clinical significance
phosphorylase, glycogen; muscle (McArdle syndrome, glycogen storage disease type V) Identifiers Symbol PYGM Entrez 5837 HUGO 9726 OMIM 608455 RefSeq NM_005609 UniProt P11217 Other data EC number 2.4.1.1 Locus Chr. 11 q12-q13.2 phosphorylase, glycogen; liver (Hers disease, glycogen storage disease type VI) Identifiers Symbol PYGL Entrez 5836 HUGO 9725 OMIM 232700 RefSeq NM_002863 UniProt P06737 Other data EC number 2.4.1.1 Locus Chr. 14 q11.2-24.3 phosphorylase, glycogen; brain Identifiers Symbol PYGB Entrez 5834 HUGO 9723 OMIM 138550 RefSeq NM_002862 UniProt P11216 Other data EC number 2.4.1.1 Locus Chr. 20 p11.2-p11.1 The inhibition of glycogen phosphorylase has been proposed as one method for treating type 2 diabetes.[10] Since glucose production in the liver has been shown to increase in type 2 diabetes patients,[11] inhibiting the release of glucose from the liver’s glycogen’s supplies appears to be a valid approach. The cloning of the human liver glycogen phosphorylase (HLGP) revealed a new allosteric binding site near the subunit interface that is not present in the rabbit muscle glycogen phosphorylase (RMGP) normally used in studies. This site was not sensitive to the same inhibitors as those at the AMP allosteric site,[12] and most success has been had synthesizing new inhibitors that mimic the structure of glucose, since glucose-6-phosphate is a known inhibitor of HLPG and stabilizes the less active T-state.[13] These glucose derivatives have had some success in inhibiting HLPG, with predicted Ki values as low as 0.016 mM.[14]
Mutations in the muscle isoform of glycogen phosphorylase (PYGM) are associated with McArdle's Disease (glycogen storage disease type V). More than 65 mutations in the PYGM gene that lead to McArdle disease have been identified to date.[15][16] Symptoms of McArdle disease include muscle weakness, myalgia, and lack of endurance, all stemming from low glucose levels in muscle tissue.[17]
Mutations in the liver isoform of glycogen phosphorylase (PYGL) are associated with Hers' Disease (glycogen storage disease type VI).[18][19] Hers' disease is often associated with mild symptoms normally limited to hypoglycemia, and is sometimes difficult to diagnose due to residual enzyme activity.[20]
The brain isoform of glycogen phosphorylase (PYGLB) has been proposed as a biomarker for gastric cancer.[21]
Regulation
Glycogen phosphorylase is regulated by both allosteric control and by phosphorylation.
Hormones such as epinephrine, insulin and glucagon regulate glycogen phosphorylase using second messenger amplification systems that are linked to G proteins. Epinephrine activates adenylate cyclase through a seven transmembrane receptor coupled to Gs which, in turn, activates adenylate cyclase to increase intracellular concentrations of cAMP. cAMP binds to and releases an active form of protein kinase A (PKA). Next, PKA phosphorylates phosphorylase kinase, which, in turn, phosphorylates glycogen phosphorylase b, transforming it into the active glycogen phosphorylase a. This phosphorylation is added onto the glycogen phosphorylase b serine 14. In the liver, glucagon activates another G-protein-linked receptor that triggers a different cascade, resulting in the activation of Phospholipase C (PLC). PLC indirectly causes the release of calcium from the hepatocytes' endoplasmic reticulum into the cytosol. The increased calcium availability binds to the calmodulin subunit and activates glycogen phosphorylase kinase. Glycogen phosphorylase kinase activates glycogen phosphorylase in the same manner mentioned previously.
Glycogen phosphorylase b is not always inactive in muscle, as it can be activated allosterically by AMP. An increase in AMP concentration, which occurs during strenuous exercise, signals energy demand. AMP activates glycogen phosphorylase b by changing its conformation from a tense to a relaxed form. This relaxed form has similar enzymatic properties as the phosphorylated enzyme. An increase in ATP concentration opposes this activation by displacing AMP from the nucleotide binding site, indicating sufficient energy stores.
Upon eating a meal, there is a release of insulin, signaling glucose availability in the blood. Insulin indirectly activates PP-1 and phosphodiesterase. The PP-1 directly dephosphorylates glycogen phosphorylase a, reforming the inactive glycogen phosphorylase b. The phosphodiesterase converts cAMP to AMP. This activity removes the second messenger (generated by glucagon and epinephrine) and inhibits PKA. In this manner, PKA can no longer cause the phosphorylation cascade that ends with formation of (active) glycogen phosphorylase a. These modifications initiated by insulin end glycogenolysis in order to preserve what glycogen stores are left in the cell and trigger glycogenesis (rebuilding of glycogen).
Phosphorylase a and phosphorylase b each exist in two forms a T (tense) inactive state and R (relaxed) state. Phosphorylase b is normally in the T state, inactive due to the physiological presence of ATP and Glucose 6 phosphate, and Phosphorylase a is normally in the R state (active).
An isoenzyme of glycogen phosphorylase exists in the liver sensitive to glucose concentration, as the liver acts as a glucose exporter. In essence, liver phosphorylase is responsive to glucose, which causes a very responsive transition from the R to T form, inactivating it; furthermore, liver phosphorylase is insensitive to AMP.
Historical significance
Glycogen phosphorylase was the first allosteric enzyme to be discovered.[7] This accomplishment was one of many landmark achievements made by Carl and Gerty Cori. In 1943, with the help of Arda Green, the pair illustrated that glycogen phosphorylase existed in either the a or b forms depending on its phosphorylation state, as well as in the R or T states based on the presences of AMP.[22]
References
- ^ PDB 3E3N
- ^ a b Livanova NB, Chebotareva NA, Eronina TB, Kurganov BI (May 2002). "Pyridoxal 5′_Phosphate as a Catalytic and Conformational Cofactor of Muscle Glycogen Phosphorylase b". Biochemistry (Moscow) 67 (10): 1089–1998. doi:10.1023/A:1020978825802. PMID 12460107.
- ^ Palm D, Klein HW, Schinzel R, Buehner M, Helmreich, EJM (February 1990). "The role of pyridoxal 5'-phosphate in glycogen phosphorylase catalysis". Biochemistry 29 (5): 1099–1107. doi:10.1021/bi00457a001. PMID 2182117.
- ^ Browner MF, Fletterick RJ (February 1992). "Phosphorylase: a biological transducer". Trends in Biochemical Science 17 (2): 66–71. doi:10.1016/0968-0004(92)90504-3. PMID 1566331.
- ^ a b Johnson LN (March 1992). "Glycogen phosphorylase: control by phosphorylation and allosteric effectors". FASEB Journal 6 (6): 2274–82. PMID 1544539.
- ^ Newgard CB, Hwang PK, Fletterick, RJ (1989). "The family of glycogen phosphorylases: structure and function". Critical Reviews Biochemistry and Molecular Biology 24 (1): 69–99. doi:10.3109/10409238909082552. PMID 2667896.
- ^ a b Johnson LN, Barford, D (February 1990). "Glycogen phosphorylase. The structural basis of the allosteric response and comparison with other allosteric proteins.". Journal of Biological Chemistry 265 (5): 2409–2412. PMID 2137445.
- ^ Meyer F, Heilmeyer LM Jr, Haschke RH, Fischer EH (Dec 1970). "Control of phosphorylase activity in a muscle glycogen particle. I. Isolation and characterization of the protein-glycogen complex". Journal of Biological Chemistry 245 (24): 6642–6648. PMID 4320610.
- ^ David ES, Crerar MM (January 1986). "Quantitation of muscle glycogen phosphorylase mRNA and enzyme amounts in adult rat tissues". Biochim. Biophys. Acta 880 (1): 78–90. PMID 3510670.
- ^ Somsák L, Nagya V, Hadady Z, Docsa T, Gergely P. (2003). "Glucose analog inhibitors of glycogen phosphorylases as potential antidiabetic agents: recent developments". Current Pharmacological Design 9 (15): 1177–89. doi:10.2174/1381612033454919. PMID 12769745.
- ^ Moller DE (Dec 2001). "New drug targets for type 2 diabetes and the metabolic syndrome". Nature 414 (6865): 821–7. doi:10.1038/414821a. PMID 11742415.
- ^ Coats WS, Browner MF, Fletterick RJ, Newgard CB (Aug 1991). "An engineered liver glycogen phosphorylase with AMP allosteric activation". Journal of Biological Chemistry 266 (24): 16113–9. PMID 1874749.
- ^ Oikonomakos NG, Kontou M, Zographos SE, Tsitoura HS, Johnson LN, Watson KA, Mitchell EP, Fleet GW, Son JC, Bichard CJ, et al. (Jul 1994). "The design of potential antidiabetic drugs: experimental investigation of a number of beta-D-glucose analogue inhibitors of glycogen phosphorylase". European Journal of Drug Metabolism and Pharmacology 19 (3): 185–92. doi:10.1007/BF03188920. PMID 7867660.
- ^ Hopfinger A J, Reaka A, Venkatarangan P, Duca J S, Wang S. (Sep 1999). "Prediction of Ligand−Receptor Binding Free Energy by 4D-QSAR Analysis: Application to a Set of Glucose Analogue Inhibitors of Glycogen Phosphorylase". Journal of Chemical Information and Computer Science 39: 1141–1150. doi:10.1021/ci9900332.
- ^ Nogales-Gadea G, Arenas J, Andreu AL (January 2007). "Molecular genetics of McArdle's disease". Curr Neurol Neurosci Rep 7 (1): 84–92. doi:10.1007/s11910-007-0026-2. PMID 17217859.
- ^ Andreu AL, Nogales-Gadea G, Cassandrini D, Arenas J, Bruno C (July 2007). "McArdle disease: molecular genetic update". Acta Myol 26 (1): 53–7. PMID 17915571.
- ^ Grünfeld JP, Ganeval D, Chanard J, Fardeau M, Dreyfus JC (Jun 1972). "Acute renal failure in McArdle's disease. Report of two cases". New England Journal of Medicine 286 (23): 1237–41. doi:10.1056/NEJM197206082862304. PMID 4502558.
- ^ Burwinkel B, Bakker HD, Herschkovitz E, Moses SW, Shin YS, Kilimann MW (April 1998). "Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI". Am. J. Hum. Genet. 62 (4): 785–91. doi:10.1086/301790. PMC 1377030. PMID 9529348. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1377030.
- ^ Chang S, Rosenberg MJ, Morton H, Francomano CA, Biesecker LG (May 1998). "Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI". Hum. Mol. Genet. 7 (5): 865–70. doi:10.1093/hmg/7.5.865. PMID 9536091.
- ^ Tang NL, Hui J, Young E, Worthington V, To KF, Cheung KL, Li CK, Fok TF (Jun 2003). "A novel mutation (G233D) in the glycogen phosphorylase gene in a patient with hepatic glycogen storage disease and residual enzyme activity". Molecular Genetics and Metabolism 79 (2): 142–145. doi:10.1016/S1096-7192(03)00068-4. PMID 12809646.
- ^ Shimada S, Matsuzaki H, Marutsuka T, Shiomori K, Ogawa M (July 2001). "Gastric and intestinal phenotypes of gastric carcinoma with reference to expression of brain (fetal)-type glycogen phosphorylase". J. Gastroenterol. 36 (7): 457–64. doi:10.1007/s005350170068. PMID 11480789.
- ^ Cori GT, Green AA (July 1943). "Crystalline muscle phosphorylase II prosthetic group". Journal of Biological Chemistry 151 (1): 21–29. http://www.jbc.org/content/151/1.toc.
Further reading
External links
- GeneReviews/NCBI/NIH/UW entry on Glycogen Storage Disease Type VI - Hers disease
- MeSH Glycogen+phosphorylase
- Diwan JJ. "Glycogen Metabolism". Molecular Biochemistry I. Rensselaer Polytechnic Institute. http://rpi.edu/dept/bcbp/molbiochem/MBWeb/mb1/part2/glycogen.htm#animat1. Retrieved 2009-01-10.
- Goodsell DS (2001-12-01). "Glycogen Phosphorylase". Molecule of the Month. RCSB Protein Data Bank. http://www.rcsb.org/pdb/static.do?p=education_discussion/molecule_of_the_month/pdb24_1.html. Retrieved 2009-01-10.
Glycogenesis Phosphoglucomutase · UDP-glucose pyrophosphorylase · Glycogen synthase · Glycogen branching enzyme
GlycogeninGlycogenolysis extralysosomal: Glycogen phosphorylase · Debranching enzyme · Phosphoglucomutase
lysosomal: Alpha-glucosidase (Acid)Regulation Transferases: glycosyltransferases (EC 2.4) 2.4.1: Hexosyl-
transferases2.4.2: Pentosyl-
transferasesOtherOther2.4.99: Sialyl
transferasesCategories:- Genes on chromosome 11
- Genes on chromosome 14
- Genes on chromosome 20
- EC 2.4.1
Wikimedia Foundation. 2010.