- Wegener's granulomatosis
-
Wegener's granulomatosis Classification and external resources 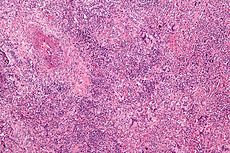
Micrograph showing features characteristic of Wegener's granulomatosis - a vasculitis and granulomas with multi-nucleated giant cells. H&E stain.ICD-10 M31.3 ICD-9 446.4 DiseasesDB 14057 MedlinePlus 000135 eMedicine med/2401 MeSH D014890 Wegener's granulomatosis (WG), more recently granulomatosis with polyangiitis (Wegener's) (GPA), is an incurable form of vasculitis (inflammation of blood vessels) that affects the nose, lungs, kidneys and other organs. Due to its end-organ damage, it is life-threatening and requires long-term immunosuppression.[1] Five-year survival is up to 87%, with some of the mortality due to toxicity of treatment. It is named after Dr. Friedrich Wegener, who described the disease in 1936.[2] In 2011, three professional bodies proposed a more descriptive name.[3]
Wegener's granulomatosis is part of a larger group of vasculitic syndromes, all of which feature an autoimmune attack by an abnormal type of circulating antibody termed ANCAs (antineutrophil cytoplasmic antibodies) against small and medium-size blood vessels. Apart from Wegener's, this category includes Churg-Strauss syndrome and microscopic polyangiitis.[1] Although Wegener's granulomatosis affects small and medium-sized vessels,[4] it is formally classified as one of the small vessel vasculitides in the Chapel Hill system.[5]
Contents
Signs and symptoms
Initial signs are extremely variable, and diagnosis can be severely delayed due to the nonspecific nature of the symptoms. Rhinitis is generally the first sign in most patients.[1]
- Kidney: rapidly progressive glomerulonephritis (75%), leading to chronic renal failure
- Upper airway, eye and ear disease:
- Nose: pain, stuffiness, nosebleeds, rhinitis, crusting, saddle-nose deformity due to a perforated septum
- Ears: conductive hearing loss due to auditory tube dysfunction, sensorineural hearing loss (unclear mechanism)
- Oral cavity: strawberry gingivitis, underlying bone destruction with loosening of teeth, non-specific ulcerations throughout oral mucosa
- Eyes: pseudotumours, scleritis, conjunctivitis, uveitis, episcleritis
- Trachea: subglottal stenosis
- Lungs: pulmonary nodules (referred to as "coin lesions"), infiltrates (often interpreted as pneumonia), cavitary lesions, pulmonary hemorrhage causing hemoptysis, and rarely bronchial stenosis.
- Arthritis: Pain or swelling (60%), often initially diagnosed as rheumatoid arthritis
- Skin: nodules on the elbow, purpura, various others (see cutaneous vasculitis)
- Nervous system: occasionally sensory neuropathy (10%) and rarely mononeuritis multiplex
- Heart, gastrointestinal tract, brain, other organs: rarely affected.
Diagnosis
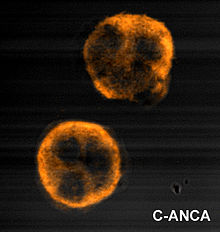 Immunofluorescence pattern produced by binding of ANCA to ethanol-fixed neutrophils, from a patient with Wegener's Granulomatosis.
Immunofluorescence pattern produced by binding of ANCA to ethanol-fixed neutrophils, from a patient with Wegener's Granulomatosis.
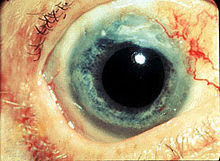 Photo showing the sclerokeratitis associated with Wegener's granulomatosis.
Photo showing the sclerokeratitis associated with Wegener's granulomatosis.Wegener's granulomatosis is usually suspected only when a patient has had unexplained symptoms for a long period of time. Determination of ANCAs can aid in the diagnosis, but positivity is not conclusive and negative ANCAs are not sufficient to reject the diagnosis. Cytoplasmic staining ANCAs that react with the enzyme proteinase 3 (cANCA) in neutrophils (a type of white blood cell) are associated with Wegener's.[1]
If the patient has renal failure or cutaneous vasculitis, these are the most logical organs to obtain a biopsy from. Rarely, thoracoscopic lung biopsy is required. On histopathological examination, a biopsy will show leukocytoclastic vasculitis with necrotic changes and granulomatous inflammation (clumps of typically arranged white blood cells) on microscopy. These granulomas are the main reason for the appellation of "Wegener's granulomatosis", although it is not an essential feature. Unfortunately, many biopsies can be nonspecific and 50% provide too little information for the diagnosis of Wegener's.[1]
Differential diagnosis (alternative possible diagnoses) can be extensive. ANCAs can be positive after the use of certain drugs and other forms of vasculitis can present with very similar symptoms. The saddle-nose deformity may also be seen in relapsing polychondritis, cocaine abuse and in congenital syphilis.
Criteria
In 1990, the American College of Rheumatology accepted classification criteria for Wegener's. These criteria were not intended for diagnosis, but for inclusion in randomised controlled trials. Two or more positive criteria have a sensitivity of 88.2% and a specificity of 92.0% of describing Wegener's.[6]
- Nasal or oral inflammation:
- painful or painless oral ulcers or
- purulent or bloody nasal discharge
- Lungs: abnormal chest X-ray with:
- nodules,
- infiltrates or
- cavities
- Kidneys: urinary sediment with:
- microhematuria or
- red cell casts
- Biopsy: granulomatous inflammation
- within the arterial wall or
- in the perivascular area
According to the Chapel Hill Consensus Conference (CHCC) on the nomenclature of systemic vasculitis (1992), establishing the diagnosis of Wegener's granulomatosis demands:[7]
- a granulomatous inflammation involving the respiratory tract, and
- a vasculitis of small to medium-size vessels.
Several investigators have compared the ACR and Chapel Hill criteria.[8]
Pathophysiology
Inflammation with granuloma formation against a nonspecific inflammatory background is the classical tissue abnormality in all organs affected by Wegener's granulomatosis.[1]
It is now widely presumed that the anti-neutrophil cytoplasmic antibodies (ANCAs) are responsible for the inflammation in Wegener's.[1] The typical ANCAs in Wegener's are those that react with proteinase 3, an enzyme prevalent in neutrophil granulocytes.[9] This type of ANCA is also known as cANCA, with the c indicating cytoplasmic (in contrast to pANCA, which is perinuclear).
In vitro studies have found that ANCAs can activate neutrophils, increase their adherence to endothelium, and induce their degranulation that can damage endothelial cells. In theory, this phenomenon could cause extensive damage to the vessel wall, particularly of arterioles.[1] However, the presence of ANCA bound to circulating neutrophils has never been found, the activation status of neutrophils from patients with high ANCA titer does not differ from normal individuals and the presence of ANCA is not always associated with development of vasculitis.
The exact cause for the production of ANCAs is unknown, although some drugs have been implicated in secondary forms of Wegener's. As with many autoimmune disorders, the cause is probably genetic predisposition combined with molecular mimicry caused by a virus or bacterium.
Treatment
Before steroid treatment became available, mortality within one year was over 90%, with average survival being 5 months. Steroids prolonged average survival to 8 months. The introduction of cyclophosphamide (CYC) in the 1970s was a major breakthrough. Five-year survival is now 87%.[10]
Initial treatment is generally with corticosteroids and oral CYC, 1 mg/kg/day and 2 mg/kg/day, respectively. Occasionally CYC is given in monthly intravenous (IV) doses. Monitoring of the white blood count is essential during CYC therapy. Once remission is attained (normally 3 to 6 months), treatment is frequently changed to azathioprine or methotrexate, which are less toxic drugs. Total duration of therapy should be at least one year, or longer in high risk patients. Corticosteroids are tapered to a low maintenance dose, 5–10 mg/day. Plasmapheresis may be beneficial in severe disease or pulmonary hemorrhage. Experience with other treatment agents is very limited.[1]
A systematic review of 84 trials examined the evidence for various treatments in Wegener's granulomatosis. Many trials include data on pooled groups of patients with Wegener's and microscopic polyangiitis. In this review, cases are divided between localized disease, non-organ threatening, generalized organ-threatening disease and severe renal vasculitis and immediately life-threatening disease.[10]
- In localized disease, treatment with the antibiotic co-trimoxazole is recommended, with steroids in case of treatment failure.[11]
- In generalized non-organ threatening disease, remission can be induced with methotrexate and steroids, where the steroid dose is reduced after a remission has been achieved and methotrexate used as maintenance.
- In case of organ-threatening disease, pulsed intravenous cyclophosphamide with steroids is recommended. Once remission has been achieved, azathioprine and steroids can be used to maintain remission.
- In severe renal vasculitis, the same regimen is used but with the addition of plasma exchange.
- In pulmonary hemorrhage, high doses of cyclophosphamide with pulsed methylprednisolone may be used, or alternatively CYC, steroids, and plasma exchange.
In severe disease not responsive to previously mentioned treatment, the review is positive about mycophenolate mofetil, 15-deoxyspergualin, anti-thymocyte globulin, rituximab and infliximab; data was less favourable for intravenous immunoglobulin (IVIG) and etanercept.[10]
In some patients with severe subglottic stenosis, tracheotomy is required to maintain an airway.
Follow-up: general well-being and laboratory organ markers are checked on a regular basis to ascertain the patient has remained in remission.
On 19 April 2011, the FDA announced its approval of rituximab in combination with glucocorticoids to treat this condition.[12]
Epidemiology
The incidence is 10 cases per million per year.[10] 90% of the patients are white. While it mainly occurs in the middle-aged, it has been reported in much younger and older patients.
Prognosis
25 to 40% of patients suffer from flare-ups, but a majority respond well to treatment. Anatomical problems (sinusitis, tracheal stenosis) may require surgery in a small proportion. Relapses can be long and troublesome.
Long-term complications are very common (86%): mainly chronic renal failure, hearing loss and deafness.[1]
History
Scottish otolaryngologist Peter McBride (1854–1946) first described the condition in 1897 in a BMJ article entitled "Photographs of a case of rapid destruction of the nose and face".[13] Heinz Karl Ernst Klinger (born 1907) would add information on the anatomical pathology, but the full picture was presented by Friedrich Wegener (1907–1990), a German pathologist, in two reports in 1936 and 1939.[2]
An earlier name for the disease was pathergic granulomatososis.[14] The disease is still sometimes confused with lethal midline granuloma and lymphomatoid granulomatosis, both malignant lymphomas.[15]
In 2006, Dr. Alexander Woywodt (Preston, United Kingdom) and Dr. Eric Matteson (Mayo Clinic, USA) investigated Dr. Wegener's past, and discovered that he was, at least at some point of his career, a follower of the Nazi regime. In addition, their data indicate that Dr. Wegener was wanted by Polish authorities and that his files were forwarded to the United Nations War Crimes Commission. Finally, Dr. Wegener worked in close proximity to the genocide machinery in Lodz. Their data raise serious concerns about Dr. Wegener's professional conduct. They suggest that the eponym be abandoned and propose "ANCA-associated granulomatous vasculitis."[16] The authors have since campaigned for other medical eponyms to be abandoned, too.[17] In 2011, the American College of Rheumatology (ACR), the American Society of Nephrology (ASN) and the European League Against Rheumatism (EULAR) resolved to change the name to granulomatosis with polyangiitis.[3]
References
- ^ a b c d e f g h i j Seo P, Stone JH (July 2004). "The antineutrophil cytoplasmic antibody-associated vasculitides". Am. J. Med. 117 (1): 39–50. doi:10.1016/j.amjmed.2004.02.030. PMID 15210387.
- ^ a b synd/2823 at Who Named It?
- ^ a b Falk RJ, Gross WL, Guillevin L et al (2011). "Granulomatosis with polyangiitis (Wegener's): An alternative name for Wegener's granulomatosis". Ann. Rheum. Dis. 70: 74. doi:10.1136/ard.2011.150714. PMID 21372195.
- ^ "Wegener's Granulomatosis: Vasculitis: Merck Manual Professional". http://www.merck.com/mmpe/sec04/ch033/ch033k.html. Retrieved 2009-01-08.
- ^ Silva, Fred; Jennette, J. Charles; Heptinstall, Robert H.; Olson, Jean T.; Schwartz, Melvin (2007). Hepinstall's pathology of the kidney. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 677. ISBN 0-7817-4750-3.
- ^ Leavitt RY, Fauci AS, Bloch DA, et al. (August 1990). "The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis". Arthritis Rheum. 33 (8): 1101–7. doi:10.1002/art.1780330807. PMID 2202308.
- ^ Jennette JC, Falk RJ, Andrassy K, et al. (February 1994). "Nomenclature of systemic vasculitides. Proposal of an international consensus conference". Arthritis Rheum. 37 (2): 187–92. doi:10.1002/art.1780370206. PMID 8129773.
- ^ Bruce IN, Bell AL (April 1997). "A comparison of two nomenclature systems for primary systemic vasculitis". Br. J. Rheumatol. 36 (4): 453–8. doi:10.1093/rheumatology/36.4.453. PMID 9159539.
- ^ van der Woude FJ, Rasmussen N, Lobatto S, et al. (February 1985). "Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis". Lancet 1 (8426): 425–9. doi:10.1016/S0140-6736(85)91147-X. PMID 2857806.
- ^ a b c d Bosch X, Guilabert A, Espinosa G, Mirapeix E (2007). "Treatment of antineutrophil cytoplasmic antibody associated vasculitis: a systematic review". JAMA 298 (6): 655–69. doi:10.1001/jama.298.6.655. PMID 17684188.
- ^ Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG (1996). "Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group". N. Engl. J. Med. 335 (1): 16–20. doi:10.1056/NEJM199607043350103. PMID 8637536. http://content.nejm.org/cgi/content/abstract/335/1/16.
- ^ "FDA approves Rituxan to treat two rare disorders" (Press release). Food and Drug Administration. 19 April 2011. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm251946.htm. Retrieved 20 April 2011.
- ^ Friedmann I (1982). "McBride and the midfacial granuloma syndrome. (The second 'McBride Lecture', Edinburgh, 1980)". The Journal of laryngology and otology 96 (1): 1–23. PMID 7057076.
- ^ Fienberg R (1955). "Pathergic granulomatosis". Am. J. Med. 19 (6): 829–31. doi:10.1016/0002-9343(55)90150-9. PMID 13275478.
- ^ Mendenhall WM, Olivier KR, Lynch JW Jr, Mendenhall NP (2006). "Lethal midline granuloma-nasal natural killer/T-cell lymphoma". Am J Clin Oncol 29 (2): 202–6. doi:10.1097/01.coc.0000198738.61238.eb. PMID 16601443.
- ^ Woywodt A, Matteson EL (2006). "Wegener's granulomatosis--probing the untold past of the man behind the eponym". Rheumatology (Oxford) 45 (10): 1303–6. doi:10.1093/rheumatology/kel258. PMID 16887845.
- ^ Woywodt A, Matteson E (2007). "Should eponyms be abandoned? Yes". BMJ 335 (7617): 424. doi:10.1136/bmj.39308.342639.AD. PMC 1962844. PMID 17762033. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1962844.
External links
- Classification criteria by the American College of Rheumatology
Vasculitis/arteritis: systemic vasculitis (M30–M31, 446) Large vessel Medium vessel Small vessel Pauci-immuneHypersensitivity vasculitis/Henoch–Schönlein purpuraUngroupedAcute hemorrhagic edema of infancy · Bullous small vessel vasculitis · Cutaneous small-vessel vasculitisOther Goodpasture's syndrome · Sneddon's syndromeUrinary system · Pathology · Urologic disease / Uropathy (N00–N39, 580–599) Abdominal Primarily
nephrotic.3 Mesangial proliferative · .4 Endocapillary proliferative .5/.6 Membranoproliferative/mesangiocapillaryBy conditionType III RPG/Pauci-immuneWegener's granulomatosis · Microscopic polyangiitisTubulopathy/
tubulitisAny/allAny/allGeneral syndromesOtherUreterPelvic UrethraUrethritis (Non-gonococcal urethritis) · Urethral syndrome · Urethral stricture/Meatal stenosis · Urethral caruncleAny/all Obstructive uropathy · Urinary tract infection · Retroperitoneal fibrosis · Urolithiasis (Bladder stone, Kidney stone, Renal colic) · Malacoplakia · Urinary incontinence (Stress, Urge, Overflow)Categories:- Arthritis
- Diseases involving the fasciae
- Rheumatology
- Nephrology
- Lung disorders
- Autoimmune diseases
- Vascular-related cutaneous conditions
- Systemic connective tissue disorders


Wikimedia Foundation. 2010.