- Discovery and development of antiandrogens
-
In the 1960s, the first antiandrogen, or androgen antagonist, was discovered. Antiandrogens antagonise the androgen receptor (AR) and thereby block the biological effects of testosterone and dihydrotestosterone (DHT). Antiandrogens are important for men with hormonally responsive diseases like prostate cancer, benign prostatic hyperplasia (BHP), acne, seborrhea, hirsutism and androgen alopecia. Antiandrogens are mainly used for the treatment of prostate diseases.[1][2][3] New research also suggests that ARs could be linked to the disease progression of triple-negative breast cancer and that antiandrogens can potentially be used to treat it.[4]
Current antiandrogens are small molecule antiandrogens and can be either steroidal or nonsteroidal depending on ligand chemistry. Steroidal antiandrogens share a similar steroid structure, while nonsteroidal antiandrogens may have structurally distinctive pharmacophores. Only a limited number of compounds are available for clinical use despite the fact that a very large variety of antiandrogen compounds have been discovered and researched.[2]
Contents
History
At the beginning of the twentieth century, a relationship between the pituitary, testes and prostate gland was established. Charles Brenton Huggins established a method to measure the effect hormone changes have on prostatic function. He found out that castration or estrogen administration led to glandular atrophy, which could be reversed by re-administration of androgen. In 1941 the beneficial effect of androgen ablation on metastatic prostate cancer was realised when Huggins and Clarence Hodges treated these patients by either castration or estrogen therapy. They monitored the prostate size and therapeutic efficacy by measuring serum acid-phosphatase levels. They concluded that androgenic activity in the body influences prostate cancer at least with respect to serum phosphatase. Huggins was the first to use a systemic approach to treat prostate cancer. He was awarded the Nobel Prize in Physiology or Medicine in 1966.[1]
It became evident after many findings that androgen ablation, by means of castration or estrogen administration, was not enough to completely cure patients with advanced prostate cancer. In the late 1960s, the AR was discovered and characterised. Screening of chemical libraries for AR blockers led to the discovery of cyproterone. An acetate group was then added to cyproterone creating cyproterone acetate. In the 1970s, flutamide was discovered. The United States Food and Drug Administration (FDA) approved it in 1989 for use in treating prostate cancer. In 1995, bicalutamide was also approved and nilutamide followed a year later.[1][5]
Androgen receptor
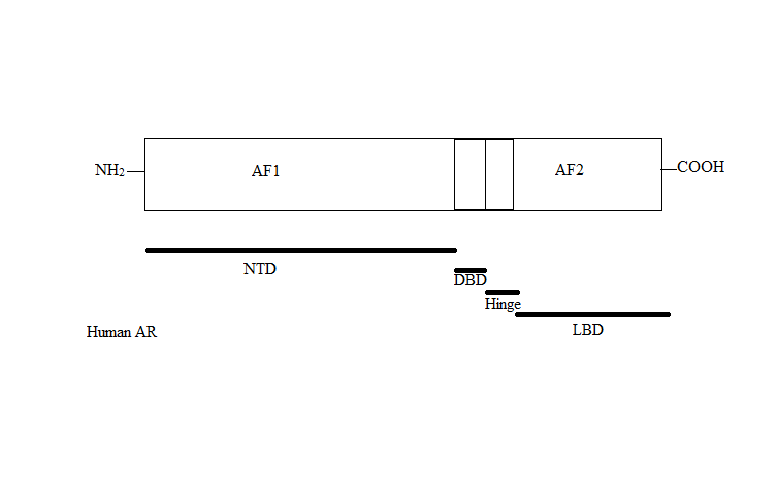
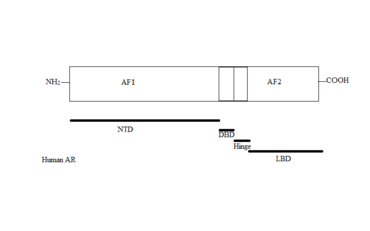 Figure 1 The four main regions of the androgen receptor
Figure 1 The four main regions of the androgen receptor
The AR belongs to the steroid receptor subfamily of the nuclear receptor superfamily. Its function is regulated by the binding of androgens, which initiates sequential conformation changes of the receptor that affects receptor-protein and receptor-DNA interactions. Endogenous androgens are mainly testosterone and DHT.[6][7][8][9]
The AR gene is more than 90kb long and codes for a protein of 919 amino acids. Only one AR gene has been identified in humans which is located on chromosome X. It comprises four main regions, see figure 1:[2][3][5][6]
- N-terminal domain (NTD) which serves a modulatory function.
- DNA-binding domain (DBD) which recognises and binds to androgen response elements (ARE) in target gene sequence.
- Ligand binding domain (LBD) which is responsible for ligand recognition and binding.
- A small hinge region between the DBD and LBD.
Two functions have been identified in AR that have critical roles in the regulation of target gene transactivation, the N-terminal activation function 1 (AF1) and the C-terminal activation function 2 (AF2). AF1 is ligand-independent and plays the primary role in target gene transactivation. The AF2 is a ligand-dependent and only shows limited function.[6][8]
Mechanism of action
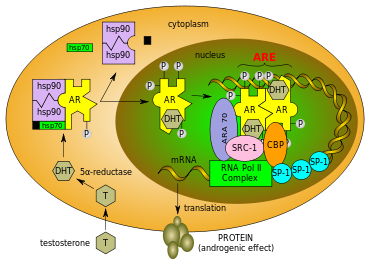 Figure 2 Mechanism of action for the androgen receptor
Figure 2 Mechanism of action for the androgen receptorUnbound AR is mainly located in the cytoplasm, like a typical steroid receptor, and is associated with a complex of heat shock proteins (HSP) through interactions with LBD. Androgens, either agonists or antagonists, position themselves in the ligand-binding pocket (LBP) of the cytosolic AR and bind to the LBD, see figure 2. The AR goes through a series of conformational changes and HSP dissociate from AR. The transformed AR undergoes dimerisation, phosphorylation and translocates to the nucleus. The translocated receptor then binds to the androgen-response elements (ARE) on the promoter of the androgen responsive gene, a consensus sequence located either upstream or downstream of the transcription start site (TSS) of AR target genes. Recruitment of other transcription co-factors (including co-activators and co-repressors) and general transcriptional machinery further ensures the transactivation of AR-regulated gene expression. All these complicated processes are initiated by the ligand-induced conformational changes in the LBD. Ligand specific recruitment of coregulators might be crucial for the agonist or antagonist activity of AR ligands. Binding of DNA is also required for AR-regulated gene expression, also known as classic genomic gene function of AR.[5][6][8]
Development of steroidal- and non-steroidal antiandrogens
Cyproterone is a steroidal antiandrogen that competitively inhibits the binding of testosterone or DHT to AR. Cyproterone binds to ARs that are expressed by prostate cancer cells as well as to the AR that are expressed in the hypothalamus and pituitary. Therefore cyproterone blocks the negative feedback of androgens at the hypothalamic-pituitary level leading to increased luteinizing hormone (LH) serum levels. This rise in LH levels causes an increase in serum testosterone levels and ultimately diminishes the ability of cyproterone to compete for AR binding and to block androgenic stimulation.[1][5]
 Figure 3 Cyproterone acetate
Figure 3 Cyproterone acetateCyproterone acetate was developed to overcome this problem. It is formed by adding an acetate group to cyproterone, see figure 3. Cyproterone acetate has a dual mode of action as it competes directly with DHT for binding to AR, but also inhibits gonadotropin secretion. It thereby reduces androgen, estrogen and LH levels.[1][5] Cyproterone acetate acts both directly as an antiandrogen in prostate cancer cells and also functions to indirectly decrease serum testosterone levels. The latter causes the limitations of cyproterone acetate, which are central effects on androgen secretion, with subsequent loss of libido and sexual potency. Several reports also state that cyproterone acetate causes liver hyperplasia. These side effects gave pharmaceutical companies the incentive to search for alternative non-steroidal "pure" antiandrogens that would not have these side effects.[1] Pure antiandrogens block the androgen receptor without exerting any agonistic or any other hormonal activity.[3]
Flutamide became the first non-steroidal antiandrogen to be tested clinically. Later the non-steroidal antiandrogens bicalutamide and nilutamide were developed. The alleged advantages of these compounds were that they did not affect libido or potency like the other centrally acting compounds under development, luteinizing-hormone-releasing hormone (LHRH) agonists and cyproterone acetate. But this theory did not prove to be true. These non-steroidal antiandrogens eventually crossed the blood-brain barrier, like cyproterone, leading to a subsequent increase in serum testosterone levels.[1]
Flutamide
 Figure 4 Flutamide
Figure 4 FlutamideFlutamide is an arylpropionamide analog with pure antiandrogenic properties, seen in figure 4. It is completely absorbed from the gastrointestinal tract after oral administration and undergoes extensive first pass metabolism to its active form, 2-hydroxyflutamide, and hydrolysis product, 3-trifluoromethyl-4-nitroaniline.[5][7][8] Hydroxyflutamide is a more potent AR antagonist than flutamide in vivo, with higher binding affinity for the AR. Hydroxyflutamide has an elimination half-life of about 8 hours in humans. Hydrolysis of the amide bond represents the major metabolic pathway for this active metabolite. By reversing the stimulatory effect of DHT on ventral prostate weight, flutamide is approximately 2-fold more potent than cyproterone acetate. Hydroxyflutamide has relatively low binding affinity to AR and is therefore generally used at high doses in order to achieve complete AR blockage in therapy.[7][10]
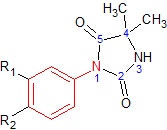
Nilutamide
 Figure 5 Nilutamide
Figure 5 NilutamideNilutamide is a nitroaromatic hydantoin analog of flutamide, as seen in figure 5.[7][8] Nilutamide is eliminated exclusively by metabolism, mainly by reduction of the aromatic nitro group. Although the hydrolysis of one of the carbonyl functions of the imidazolinedione was identified, it is much less susceptible to hepatic metabolism than the amide bond in hydroxuflutamide. This results in a longer half-life of nilutamide in humans of 2 days. Nevertheless, the nitro anion-free radical formed during nitro reduction could still be associated with hepatotoxicity in humans, especially when using relatively high dosage employed for androgen blockage.[7] Nilutamide causes side-effects which limit its usage, such as pneumonitis and delayed adaption to darkness.[5]
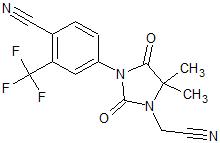
Bicalutamide
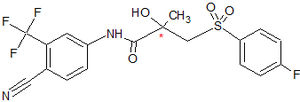 Figure 6 Bicalutamide
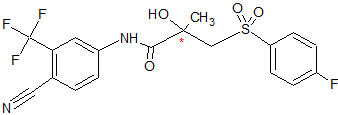
Figure 6 BicalutamideBicalutamide is an arylpropionamide analog, seen in figure 6.[7][8] It has replaced flutamide and nilutamide as the first choice antiandrogen for prostate cancer treatment. Bicalutamide is not as hepatotoxic as flutamide and nilutamide and has a longer half-life, of 6 days in humans, that allows once a day administration at lower dosage. Bicalutamide shares the amide bond structure with flutamide. Even so, the amide bond hydrolysis was discovered in rats, not in humans, which could explain the prolonged half life of bicalutamide in humans.[7]
Bicalutamide has a cyano group at the para position instead of a nitro group like flutamide and nilutamide. This change in groups avoids the nitro reduction observed in nilutamide. Bicalutamide has a chiral carbon in its structure (labeled with an asterisk in figure 6), which is connected to the hydroxyl and methyl groups . It is therefore administered as a racemate.[7] Post-approval investigation revealed that its antiandrogenic activity resides almost entirely in the (R)-enantiomer. (R)-bicalutamide has an almost fourfold higher affinity for the prostate AR than hydroxyflutamide and has a better side-effect profile compared to other antiandrogens.[7][8]
Structure and activity relationship
Steroidal antiandrogens
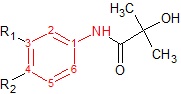
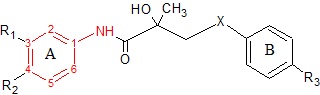
Cyproterone acetate is a 6-chloro-1,2-methylene derivative of 17 α-acetoxyprogesterone. It shows major antiandrogenic activity together with androgenic activities. Cyproterone acetate displays high affinity for AR in rats which increases when the 1,2-methylene group is removed from the compound. If the chlorine atome is replaced by a methyl group the binding slightly decreases, whereas further removal of the C6 double bond modifies the binding kinetics, see figure 7.[3]

Cyproterone acetate 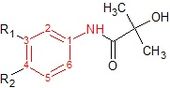
Hydroxyflutamide 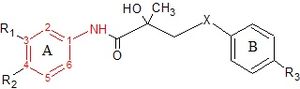
Bicalutamide 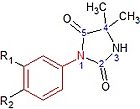
Nilutamide Figure 7: Basic structures of current antiandrogensNon-steroidal antiandrogens
Hydroxyflutamide and it‘s analogs, bicalutamide and nilutamide, share an anilide ring structure. The structures can be seen in figure 7, where the anilide ring is coloured red. These three compounds require an electron-deficient aromatic ring for efficient AR binding. Replacing the anilide with an alkene gives weakly active compounds, which can be attributed to the lack of intramolecular hydrogen binding or to poor hydrogen-bond donor capability.[3] Various combinations of electron-withdrawing substitutions in the aniline ring of these drugs, have not shown higher binding to the AR receptor than compounds which have a chloro or trifluoromethyl group at the meta position (R1) and either a cyano or nitrogen group at the para position (R2).[3][11]
For hydroxyflutamide, a group of compounds that differed in the aromatic ring did not bind to the AR. This suggests that the bisubstitution in the hydroxyflutamide ring is essential for high AR binding affinity. It has also been demonstrated that hydroxyflutamide requires the strong hydrogen bond donor ability of the tertiary hydroxyl group and fixed conformers involved in intramolecular hydrogen binding, to bind effectively to AR.[3][11]
For bicalutamide, the antiandrogenic activities of sulfide and sulfone substitions of the X-linkage were tested in vitro. The sulfides showed in most cases at least 2-fold higher binding affinity than corresponding sulfones. However, this relationship was reversed when the R3 group was NHSO2CH3, where the binding affinity of sulfone was 3-fold higher than that of sulfide. These results indicate that substituents of the B-ring largely determine the effect of the X-linkage in AR binding. Researchers have proposed that the tertiary hydroxyl group is involved in direct interaction with AR because when an acetyl group is introduced to that hydroxyl moiety, the receptor binding affinity greatly decreases.[11]
Nilutamide has very low affinity for AR when tested on castrated rat prostate. Modifications such as replacing the N3 atom with oxygen has little effect on affinity of the compound for prostate AR. By replacing the oxygen atom with a sulfur atom at the C2 position of the imidazole ring and adding butylalcohol to the N3 atom, the receptor binding and biological activity of the compound increases 100 times that of non-steroidal antiandrogens. Also the compound does not bind to other steroid receptors. If a methyl group is changed for the butylalcohol group, the compound shows 3 and 10 times more antiandrogenic activity in vivo than bicalutamide and nilutamide, respectively.[3]
Antiandrogen withdrawal syndrome
Antiandrogens that are currently on the market are particularly useful for the treatment of prostate cancer during the early stages. However, prostate cancer often progresses to a hormone-refractory state in which the cancer progresses in the presence of continued androgen ablation or antiandrogen therapy.[7] This suggests that long term use of these antiandrogens during prostate cancer can lead to the development of androgen-independent prostate cancer cells or the ability of adrenal androgens to support tumor growth.[6] This phenomenon is called antiandrogen withdrawal syndrome (AWS) and is one of the major drawbacks of existing antiandrogens. AWS is defined as tumor regression or symptomatic relief observed upon discontinuation of the antiandrogen therapy. The mechanism for this is not fully understood but current theories include alterations of the AR gene, coregulator proteins and/or signal transduction pathways. This antiandrogen resistance may also be linked to the relative weakness of current antiandrogens as they have an affinity 50 times or more lower than that of DHT for the AR. This may also explain why compensatory AR overexpression is often observed.[5]
Androgen receptor gene mutations
AR gene mutations in the LBD that alter ligand specificity and/or functional activity exist and are thought to contribute to the conversion of some AR antagonists into agonists, which explains the paradoxical temporary improvement sometimes observed in patients when antiandrogen therapy is stopped.[12] These mutations can have great affect on the antagonist activities of current small molecule antiandrogens and make them less efficient in blocking AR function via indirect modulation from inside of the LBP. Recent studies with circulating tumor cells, suggest that the mutation frequency is higher than previously assumed based on tumor biopsies.[13] The T877A,[14] W741L and W741C mutations [15] are examples of known AR LBD mutations. The LNCaP prostate cancer cell line expresses AR with a T877A point mutation that causes proliferation in the presence of the antiandrogens hydroxyflutamide and cyproterone acetate. This mutation has also been discovered in patients with antiandrogen withdrawal syndrome being treated with these compounds.[14] In another study, bicalutamide treatment of LNCaP cells resulted in two LBD mutations, W741L and W741C,[15] causing bicalutamide to acquire agonist activity to both mutant ARs.[16] The W741L mutation generates additional space such that the sulfonyl-linked phenyl ring of bicalutamide is accommodated at the location of the missing indole ring of W741.[17] In non-mutant AR, the presence of the W741 side chain probably forces bicalutamide to protrude out thus precluding the active position of H12 on the AR receptor. However, hydroxyflutamide worked as an antagonist for W741 mutant ARs.[15] This concurs with the theory that flutamide and nilutamide antagonize AR through the mechanism of “passive antagonism”, as they are of a more modest size then bicalutamide.[17] These drugs may therefore be effective as a second-line therapy for refractory prostate cancer previously treated with bicalutamide.[15]
Current status
Peptide antiandrogens
Peptide antiandrogens have been proposed to overcome the limitations of current antiandrogens regarding mutant ARs, by directly blocking AR function from protein surface, outside of the LBP. This direct blockade is thought to provide a more efficient strategy to avoid or overcome abnormal AR action during AWS, as well as allowing for more flexibility in structural modification without the space limitations of the rigid LBP.[6]
Steroid receptors have similarities in terms of gene sequences and protein structures, leading often to functional crosstalk among steroid receptors. One of the criteria for AR peptide antagonists is to achieve high degree of specificity for the AR. It is though important to realize that AR specificity does not necessarily translate in vivo, since peptide antagonists may also interact with protein targets other than AR.[6]
Ligand binding domain as target site
AR activation requires the formation of a functional activation function 2 (AF2) region in AR LBD that mediates the interactions between AR and various transcription cofactors. Therefore most of the research on peptide AR antagonists focuses on peptides that may directly block the AF2 in AR LBD from protein surface. Even in bound mutant AR, peptide antagonists would be able to block the AF2 function via direct surface interaction, regardless of the ligand bound.[6]
Research on these peptide antagonists are usually carried out by affinity screening of phage display libraries that express random peptides containing various signature motifs. ARs seem to have a distinct preference for ‘FxxLF’ type of binding motifs (where F = phenylalanine, L = leucine, and X = any amino acid residue), whereas other nuclear receptors have a highly similar binding mechanism for ‘LxxLL’ type of binding motifs. This provides a unique opportunity for the development of AR-specific peptides.[6]
Even though small molecule antagonists and peptide antagonist targeting AF2 surface differ in binding sites, they both inhibit AR function by disrupting AF2 function. Therefore mechanistically, these peptide antagonists may also be classified as ‘AF2 antagonists’.[6]
 Figure 8 Sintokamide A
Figure 8 Sintokamide AN-terminal domain as target site
Functionally, AR N-terminal domain (NTD) plays the primary role in regulating target gene transcription activation and mediating various receptor-protein and intra-receptor N-terminal and C-terminal interactions. Therefore modulation of NTD function is considered an efficient strategy to target AR action. Among various functional domains in different nuclear receptors, NTD is the least conserved and so could maybe become the best target site for peptide antagonists to achieve AR specificity. However the structural features of the NTD are undetermined due to a high degree of flexibility in its conformation. Both biochemical and circular dichroism spectroscopy analysis suggest that AR NTD is highly disordered under native conditions, making it a difficult target for drug discovery.[6]
In 2008 there were reports of a chlorinated peptide, Sintokamide A, isolated from marine sponges that effectively inhibits AR N-terminal domain-activated reporter gene transcription, see figure 8.[18] The evidence presented was not sufficient enough to support the conclusion that Sintokamide A directly inhibits the function of AR NTD, and the mechanism of action needs further investigation.[6]
Selective androgen receptor modulators
Small molecule antiandrogens that are available today have undesirable side effects caused by complete, non-selective inhibition of AR action. To minimize these side effects, a new class of tissue selective androgen receptor modulators (SARMs) has been proposed as a novel approach for the treatment of prostate cancer. These ligands should behave as antagonists in the prostate with either no activity or agonist activity in other target tissues, so as to have little or no effects in the anabolic tissues or central nervous system (CNS). However discovering this new class of ligands might be challenging because the molecular mechanism of AR action is not well understood.[6]
Several mechanisms have been proposed to achieve this tissue selectivity of AR ligands. The most definitive evidence exists for the role of 5-alpha reductase. 5-alpha reductase is only expressed in specific tissues and could therefore be a unique contributor to tissue selectivity. Specific inhibition of the type 2 enzyme by finasteride blocks the conversion of testosterone to DHT in the prostate.[6] Several approaches might make use of the potential tissue-specific conversion to develop SARMs, including:
- Inactive parent compounds that are activated by type 2 5-alpha reductase in the prostate to form antiandrogens.
- AR agonists that are inactivated by type 2 5-alpha reductase in the prostate.
- AR agonists that are converted to antiandrogens only by type 2 5-alpha reductase in the prostate.[19]
Other small molecule antiandrogens
The development status of other small molecule antiandrogens undergoing research in 2011 can be seen in table 1.
Table 1 Name of compound Structure Company Stage of development Other information RU58642 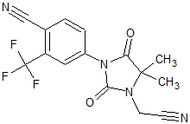
Roussel-Uclaf SA Preclinical – no further developments since 1998 Orally active and more potent than current small molecular antiandrogens.[20] LG120907 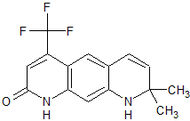
Ligand Pharmaceuticals Preclinical Orally active, strong antagonistic activity in the prostate without raising plasma levels of LH and testosterone.[21] LG105 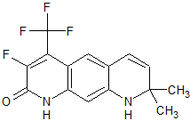
Ligand Pharmaceuticals Preclinical Orally available, strong antagonistic activity in the prostate without raising plasma levels of LH and testosterone. Seems to be more potent than LG120907.[21] RD162 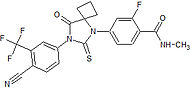
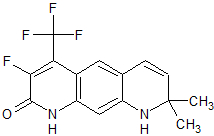
Medivation Preclinical High binding affinity to AR. Unlike bicalutamide, it does not promote nuclear translocation and impairs both DNA binding to androgen response elements and recruitment of coactivators.[22] MDV3100 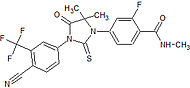
Medivation Phase III clinical High binding affinity to AR. Unlike bicalutamide, it does not promote nuclear translocation and impairs both DNA binding to androgen response elements and recruitment of coactivators.[22] Induces tumor cell apoptosis and has not agonist activity.[23] BMS-641988 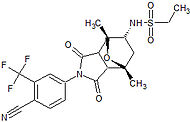
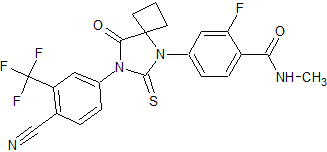
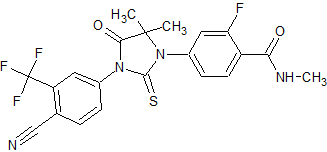
Bristol-Myers Squibb Phase I clinical – trial terminated Showed increased potency compared to bicalutamide. Phase I trial was discontinued because of an epileptic seizure in a patient.[24] Led to the findings that several antiandrogens produce off-target antagonist binding to GABA-A receptors.[25] CH5137291 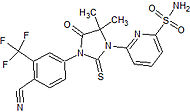
Chugai Pharmaceutical Co. Ltd. Preclinical Completely inhibits AR-mediated transactivation and proliferation of the CRPC xenograft model LNCaP-BC2, which is bicalutamide-resistant.[26][27]  Figure 9 Ataric acid
Figure 9 Ataric acid Figure 10 N-butylbenzenesulfonamide
Figure 10 N-butylbenzenesulfonamideNatural antiandrogens
Atraric acid and N-butylbenzenesulfonamide are natural compounds with antiandrogen properties which have been purified from the bark of the African tree Pygeum africanum, see figures 9 and 10.[28] In vitro assays have shown them both to be selective AR agonists and that they inhibit proliferation of several prostate cancer cell lines. Atraric acid also hinders extracellular matrix invasion and both compounds are able to prevent androgen-induced nuclear translocation of the AR. More potent derivatives are currently being synthesized in hope of improving the pharmacological profile of these two compounds.[29]
See also
- Antiandrogen
- Androgen receptor
- Prostate cancer
- Cyproterone
- Flutamide
- Bicalutamide
- Nilutamide
- Selective androgen receptor modulator
References
- ^ a b c d e f g Denmeade, SR; Isaacs, JT (2002 May). "A history of prostate cancer treatment.". Nature reviews. Cancer 2 (5): 389–96. doi:10.1038/nrc801. PMID 12044015.
- ^ a b c Gao, W (2010-10-30). "Androgen receptor as a therapeutic target.". Advanced drug delivery reviews 62 (13): 1277–84. PMID 20708648.
- ^ a b c d e f g h Singh, SM; Gauthier, S, Labrie, F (2000 Feb). "Androgen receptor antagonists (antiandrogens): structure-activity relationships.". Current medicinal chemistry 7 (2): 211–47. PMID 10637363.
- ^ Gucalp, A; Traina, TA (2010 Jan-Feb). "Triple-negative breast cancer: role of the androgen receptor.". Cancer journal (Sudbury, Mass.) 16 (1): 62–5. PMID 20164692.
- ^ a b c d e f g h Haendler, B; Cleve, A (2011-06-16). "Recent developments in antiandrogens and selective androgen receptor modulators.". Molecular and cellular endocrinology. PMID 21704118.
- ^ a b c d e f g h i j k l m n Gao, W (2010). "Peptide antagonist of the androgen receptor.". Current pharmaceutical design 16 (9): 1106–13. PMID 20030610.
- ^ a b c d e f g h i j Gao, W; Kim, J, Dalton, JT (2006 Aug). "Pharmacokinetics and pharmacodynamics of nonsteroidal androgen receptor ligands.". Pharmaceutical research 23 (8): 1641–58. PMID 16841196.
- ^ a b c d e f g Lemke, David A. Williams, Thomas L. (2002). Foye's principles of medicinal chemistry (5th ed. ed.). Baltimore [etc.]: Williams & Wilkins. ISBN 0683307371.
- ^ Narayanan, R; Mohler, ML, Bohl, CE, Miller, DD, Dalton, JT (2008). "Selective androgen receptor modulators in preclinical and clinical development.". Nuclear receptor signaling 6: e010. PMID 19079612.
- ^ Poyet, P; Labrie, F (1985 Oct). "Comparison of the antiandrogenic/androgenic activities of flutamide, cyproterone acetate and megestrol acetate.". Molecular and cellular endocrinology 42 (3): 283–8. PMID 3930312.
- ^ a b c Yin, D; He, Y, Perera, MA, Hong, SS, Marhefka, C, Stourman, N, Kirkovsky, L, Miller, DD, Dalton, JT (2003 Jan). "Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor.". Molecular pharmacology 63 (1): 211–23. PMID 12488554.
- ^ Miyamoto, H; Rahman, MM, Chang, C (2004-01-01). "Molecular basis for the antiandrogen withdrawal syndrome.". Journal of cellular biochemistry 91 (1): 3–12. PMID 14689576.
- ^ Jiang, Y; Palma, JF, Agus, DB, Wang, Y, Gross, ME (2010 Sep). "Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer.". Clinical chemistry 56 (9): 1492–5. PMID 20581083.
- ^ a b Suzuki, H; Akakura, K, Komiya, A, Aida, S, Akimoto, S, Shimazaki, J (1996 Sep). "Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: relation to antiandrogen withdrawal syndrome.". The Prostate 29 (3): 153–8. PMID 8827083.
- ^ a b c d Hara, T; Miyazaki, J, Araki, H, Yamaoka, M, Kanzaki, N, Kusaka, M, Miyamoto, M (2003-01-01). "Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome.". Cancer research 63 (1): 149–53. PMID 12517791.
- ^ Bohl, CE; Gao, W, Miller, DD, Bell, CE, Dalton, JT (2005-04-26). "Structural basis for antagonism and resistance of bicalutamide in prostate cancer.". Proceedings of the National Academy of Sciences of the United States of America 102 (17): 6201–6. PMID 15833816.
- ^ a b Nahleh, Z (2008). "Functional and structural analysis of androgen receptors for anti-cancer drug discovery". Cancer Therapy 6: 439–444. http://www.cancer-therapy.org/CT/v6/B/PDF/48._Nahleh,_439-444.pdf.
- ^ Sadar, MD; Williams, DE, Mawji, NR, Patrick, BO, Wikanta, T, Chasanah, E, Irianto, HE, Soest, RV, Andersen, RJ (2008-11-06). "Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells.". Organic letters 10 (21): 4947–50. PMID 18834139.
- ^ Gao, W; Dalton, JT (2007 Mar). "Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs).". Drug discovery today 12 (5-6): 241–8. doi:10.1016/j.drudis.2007.01.003. PMC 2072879. PMID 17331889. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2072879.
- ^ Battmann, T; Branche, C, Bouchoux, F, Cerede, E, Philibert, D, Goubet, F, Teutsch, G, Gaillard-Kelly, M (1998 Jan). "Pharmacological profile of RU 58642, a potent systemic antiandrogen for the treatment of androgen-dependent disorders.". The Journal of steroid biochemistry and molecular biology 64 (1-2): 103–11. PMID 9569015.
- ^ a b Hamann, LG; Higuchi, RI, Zhi, L, Edwards, JP, Wang, XN, Marschke, KB, Kong, JW, Farmer, LJ, Jones, TK (1998-02-12). "Synthesis and biological activity of a novel series of nonsteroidal, peripherally selective androgen receptor antagonists derived from 1,2-dihydropyridono[5,6-g]quinolines.". Journal of medicinal chemistry 41 (4): 623–39. PMID 9484511.
- ^ a b Tran, C; Ouk, S, Clegg, NJ, Chen, Y, Watson, PA, Arora, V, Wongvipat, J, Smith-Jones, PM, Yoo, D, Kwon, A, Wasielewska, T, Welsbie, D, Chen, CD, Higano, CS, Beer, TM, Hung, DT, Scher, HI, Jung, ME, Sawyers, CL (2009-05-08). "Development of a second-generation antiandrogen for treatment of advanced prostate cancer.". Science 324 (5928): 787–90. doi:10.1126/science.1168175. PMC 2981508. PMID 19359544. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2981508.
- ^ Scher, HI; Beer, TM, Higano, CS, Anand, A, Taplin, ME, Efstathiou, E, Rathkopf, D, Shelkey, J, Yu, EY, Alumkal, J, Hung, D, Hirmand, M, Seely, L, Morris, MJ, Danila, DC, Humm, J, Larson, S, Fleisher, M, Sawyers, CL, Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials, Consortium (2010-04-24). "Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study.". Lancet 375 (9724): 1437–46. PMID 20398925.
- ^ Rathkopf, D; Liu, G, Carducci, MA, Eisenberger, MA, Anand, A, Morris, MJ, Slovin, SF, Sasaki, Y, Takahashi, S, Ozono, S, Fung, NK, Cheng, S, Gan, J, Gottardis, M, Obermeier, MT, Reddy, J, Zhang, S, Vakkalagadda, BJ, Alland, L, Wilding, G, Scher, HI, Prostate Cancer Clinical Trials, Consortium (2011-02-15). "Phase I dose-escalation study of the novel antiandrogen BMS-641988 in patients with castration-resistant prostate cancer.". Clinical cancer research : an official journal of the American Association for Cancer Research 17 (4): 880–7. PMID 21131556.
- ^ Foster, WR; Car, BD, Shi, H, Levesque, PC, Obermeier, MT, Gan, J, Arezzo, JC, Powlin, SS, Dinchuk, JE, Balog, A, Salvati, ME, Attar, RM, Gottardis, MM (2011 Apr). "Drug safety is a barrier to the discovery and development of new androgen receptor antagonists.". The Prostate 71 (5): 480–8. PMID 20878947.
- ^ Kawata, H; Arai, S, Nakagawa, T, Ishikura, N, Nishimoto, A, Yoshino, H, Shiraishi, T, Tachibana, K, Nakamura, R, Sato, H (2011 Sep). "Biological properties of androgen receptor pure antagonist for treatment of castration-resistant prostate cancer: optimization from lead compound to CH5137291.". The Prostate 71 (12): 1344–56. PMID 21308717.
- ^ Yoshino, H; Sato, H, Shiraishi, T, Tachibana, K, Emura, T, Honma, A, Ishikura, N, Tsunenari, T, Watanabe, M, Nishimoto, A, Nakamura, R, Nakagawa, T, Ohta, M, Takata, N, Furumoto, K, Kimura, K, Kawata, H (2010-12-01). "Design and synthesis of an androgen receptor pure antagonist (CH5137291) for the treatment of castration-resistant prostate cancer.". Bioorganic & medicinal chemistry 18 (23): 8150–7. PMID 21050768.
- ^ Schleich, S; Papaioannou, M, Baniahmad, A, Matusch, R (2006 Jul). "Extracts from Pygeum africanum and other ethnobotanical species with antiandrogenic activity.". Planta medica 72 (9): 807–13. PMID 16783690.
- ^ Roell, D; Baniahmad, A (2011-01-30). "The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth.". Molecular and cellular endocrinology 332 (1-2): 1–8. PMID 20965230.
External links
Drug design steps in design Case studies of discovery and development of drug classes triptans · cephalosporins · antiandrogens · Bcr-Abl tyrosine kinase inhibitors · nucleoside and nucleotide reverse transcriptase inhibitors · melatonin receptor agonists · proton pump inhibitors · angiotensin receptor blockers · dual serotonin and norepinephrine reuptake inhibitors · cyclooxygenase 2 inhibitors · TRPV1 antagonistsCategories:- Antiandrogens
- Cancer research



Wikimedia Foundation. 2010.