- Discovery and development of nucleoside and nucleotide reverse-transcriptase inhibitors
-
Discovery and development of nucleoside and nucleotide reverse-transcriptase inhibitors (NRTIs and NtRTIs) began in the 1980s when the AIDS epidemic hit Western societies. NRTIs inhibit the reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of the human immunodeficiency virus (HIV). The first NRTI was zidovudine, approved by the U.S. Food and Drug Administration (FDA) in 1987, which was the first step towards treatment of HIV. Six NRTI agents and one NtRTI have followed. The NRTIs and the NtRTI are analogues of endogenous 2´-deoxy-nucleoside and nucleotide. Drug-resistant viruses are an inevitable consequence of prolonged exposure of HIV-1 to anti-HIV drugs.
Contents
History
In the summer of 1981 the acquired immunodeficiency syndrome (AIDS) was first reported.[1] Two years later the etiological link to AIDS, the human immunodeficiency virus (HIV) was identified.[2][3] Since the identification of HIV the development of effective antiretroviral drugs and the scientific achievements in HIV research has been enormous.[3][4] Antiretroviral drugs for the treatment of HIV infections belong to six categories: Nucleoside and nucleotide reverse-transcriptase inhibitors, Non-nucleoside reverse-transcriptase inhibitors, protease inhibitors, entry inhibitors, co-receptor inhibitors and integrase inhibitors.[4] The reverse transcriptase of HIV-1 has been the main foundation for the development of anti-HIV drugs.[5] The first nucleoside reverse-transcriptase inhibitor with in vitro anti-HIV activity was zidovudine.[6] Since zidovudine was approved in 1987, six nucleosides and one nucleotide reverse-transcriptase inhibitor (NRTI) have been approved by FDA.[6] NRTIs approved by the FDA are zidovudine, didanosine, zalcitabine, stavudine, lamivudine, abacavir and emtricitabine and the only nucleotide reverse-transcriptase inhibitor (NtRTI) approved is tenofovir (see table 4).[4][6]
The HIV-1 reverse transcriptase enzyme
Function
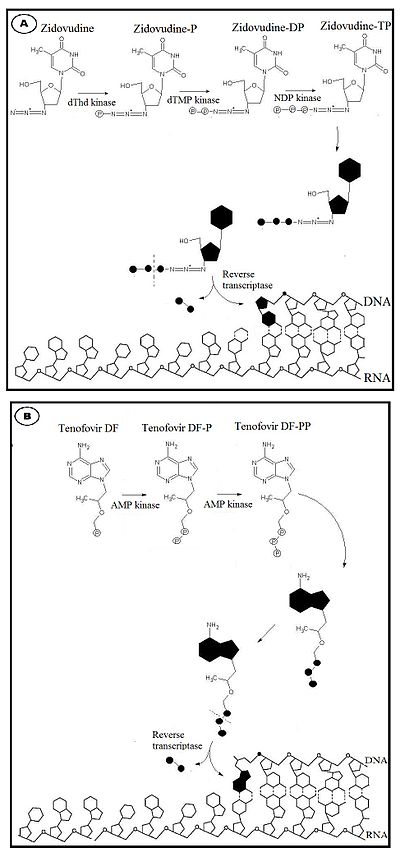 Figure 1 A: Mechanism of action of nucleoside analogues reverse transriptase inhibitors, for example zidovudine. B: Mechanism of action of the nucleotide analogue reverse-transcriptase inhibitor, tenofovir
Figure 1 A: Mechanism of action of nucleoside analogues reverse transriptase inhibitors, for example zidovudine. B: Mechanism of action of the nucleotide analogue reverse-transcriptase inhibitor, tenofovir
Most standard HIV drug thearapies revolve around inhibiting the reverse transcriptase enzyme (RT), an enzyme that is necessary to the HIV-1 virus and other retroviruses to complete their life cycle.[5] The RT enzyme serves two key functions. First, it controls the replication of the viruses genetic material via its polymerase activity. It converts the viral single-stranded RNA into an integration competent double stranded DNA. Subsequently the generated DNA is translocated into the nucleus of the host cell where it is integrated in its genome by the retroviral integrase. The other role of the RT is its ribonuclease H activity that degrades RNA only when it is in a heteroduplex with DNA.[7][8]
Structure
HIV-1 RT is an asymmetric[disambiguation needed
 ] heterodimer which is 1000 amino acid long and is composed of two subunits. The larger subunit, p66, is 560 amino acid long and it exhibits all the enzymatic activities of the RT.[8]The smaller subunit, called p51, is 440 amino acid long and it is considered to stabilizes the heterodimer but also it may take part in the binding of the tRNA primer. The p66 subunit has the two active sites: polymerase and ribonuclease H. The polymerase has four subdomains that have been named “fingers“, “thumb“, “connection“ and “palm“ for it has been compared to the right hand.[7][8][9]
] heterodimer which is 1000 amino acid long and is composed of two subunits. The larger subunit, p66, is 560 amino acid long and it exhibits all the enzymatic activities of the RT.[8]The smaller subunit, called p51, is 440 amino acid long and it is considered to stabilizes the heterodimer but also it may take part in the binding of the tRNA primer. The p66 subunit has the two active sites: polymerase and ribonuclease H. The polymerase has four subdomains that have been named “fingers“, “thumb“, “connection“ and “palm“ for it has been compared to the right hand.[7][8][9]Mechanism of action
Activation of nucleoside and nucleotide reverse-transcriptase inhibitors is primarily dependent on cellular entry by passive diffusion or carrier-mediated transport. NRTIs are highly hydrophilic and have limited membrane permeability and therefore this step is very important. NRTIs are analogues of endogenous 2´-deoxy-nucleoside and nucleotide. They are inactive in their parent forms and require successive phosphorylation.[6]
Nucleosides must be triphosphorylated, while nucleotides, which possess one phosphonated group, must be diphosphorylated.[10] This stepwise activation process occurs inside the cell and is mediated by a coordinated series of enzymes.[11] The first, and often rate limiting, phosphorylation step (for nucleoside analogues) are most commonly catalyzed by deoxynucleoside kinases. Addition of the second phosphate group to nucleoside monophosphate analogues is completed by the nucleoside monophosphate kinases (NMP kinases). A variety of enzymes are able to catalyze the final phosphorylation step for NRTIs, including nucleoside diphosphate kinase (NDP kinase), phosphoglycerate kinase, pyruvate kinase and creatine kinase, resulting in formation of respective antivirally active triphosphate analogues.[6] In their respective triphosphate forms, NRTIs and the only NtRTI available compete with their corresponding endogenous deoxynucleotide triphosphate (dNTPs) for incorporation into the nascent DNA chain (see figure 1).[6] Unlike dNTPs substrate, NRTIs lack a 3´-hydroxyl group on the deoxyribose moiety. Once incorporated into the DNA chain, the absence of a 3´-hydroxyl group, which normally forms the 5´- to 3´- phosphoester bond with the next nucleic acid, blocks further extension of the DNA by RT, and they act as chain terminators.[10][12]
Discovery and development
First step towards treatment of HIV- zidovudine
In 1964 zidovudine (AZT) was synthesized by Horwitz at the Michigan Cancer Foundation. The 3´hydroxyl group in the deoxyribose ring of thymidine is replaced by an azido group which gives us zidovudine.[13] The lack of the 3´hydroxyl group which provides the attachment point for the next nucleotide in the growing DNA chain during the reverse transcription makes it an obligate chain terminator. Ziduvodine is incorporated in place of thymidine and is an extremely potent inhibitor of HIV replication.[14] This compound had been prepared in 1964 as a potential anti-cancer agent but was shown to be ineffective.[15] In 1974 zidovudine was reported to have activity against retroviruses and was subsequently re-screened as an antiviral when the AIDS epidemic hit Western societies during mid 1980‘s.[13][15] However, ziduvodine is relatively toxic since it is converted into the triphosphate by the cellular enzymes and therefore it is activated in uninfected cells.[14]
Further development of nucleoside analogues
Dideoxynucleosides
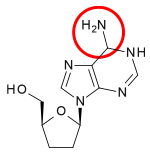
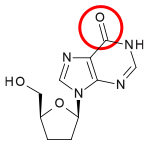
Table 1 Comparison of chemical structures: Dideoxyadenosine and didanosine Dideoxyadenosine Didanosine Chemical structure
Dideoxynucleosides are analogues of nucleoside where the sugar ring lacks both 2´ and 3´-hydroxyl groups.[9] Three years after the synthesis[disambiguation needed
] of zidovudine, Jerome Horwitz and his colleagues in Chicago prepared another dideoxynucleoside now known as zalcitabine (ddC).[16] Zalcitabine is a synthetic pyrimidine nucleoside analogue, structurally related to deoxycytidine, in which the 3´-hydroxyl group of the ribose sugar moiety is substituted with hydrogen.[17] Zalcitabine was approved by the FDA for the treatment of HIV-1 in June 1992.[3][18]2´,3´-dideoxyinosine or didanosine is converted into dideoxyadenosine in vivo. Its development has a long history.[19] In 1964 dideoxyadenosine, the corresponding adenosine analogue of zalcitabine was synthesised. Dideoxyadenosine caused kidney damage so didanosine was prepared from dideoxyadenosine by enzymatic oxidation (see table 1). It was found to be active against HIV without causing kidney damage.[16] Didanosine was approved by the FDA for the treatment of HIV-1 in October 1991.[18] Zalcitabine and didanosine are both obligate chain terminators, that have been developed for anti-HIV treatment. Unfortunately, both drugs lack selectivity[disambiguation needed
] and therefore cause side-effects.[14]Further modification of the dideoxy framework led to the development of 2´,3´-didehydro-3´-deoxythymidine (stavudine, d4T). Activity of stavudine was shown to be similar to that of zidovudine, although their phosphorylation patterns differ; the affinity[disambiguation needed
] for zidovudine to thymidine kinase (the enzyme responsible for the first phosphorylation) is similar to that of thymidine, whereas the affinity for stavudine is 700-fold weaker.[9]Table 2 Comparison of chemical structures : Zalcitabine and lamivudine Zalcitabine Lamivudine Chemical structure
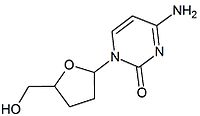

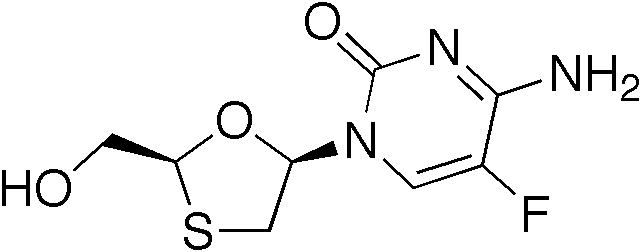
2',3'‐dideoxy‐3'‐thiacytidine (lamivudine, 3TC) was discovered by Bernard Belleau. The history of lamivudine can be traced back to the mid‐seventies while Bernard Belleau was investigating sugar derivatives. Lamivudine was developed as the sulfur analogue of zalcitabine (see table 2).[16] It was initially synthesized as a racemic mixture (BCH-189) and analysis showed that both positive and negative enantiomers of BCH-189 had in vitro activity against HIV. Lamivudine which is the negative enantiomer of 2',3'‐dideoxy‐3'‐thiacytidine and is a pyrimidine nucleoside analogue. The 3' carbon of the ribose ring of 2'-deoxycytidine has been replaced by a sulfur atom because it had greater anti-HIV activity and is less toxic than the positive enantiomer.[16][20][21]
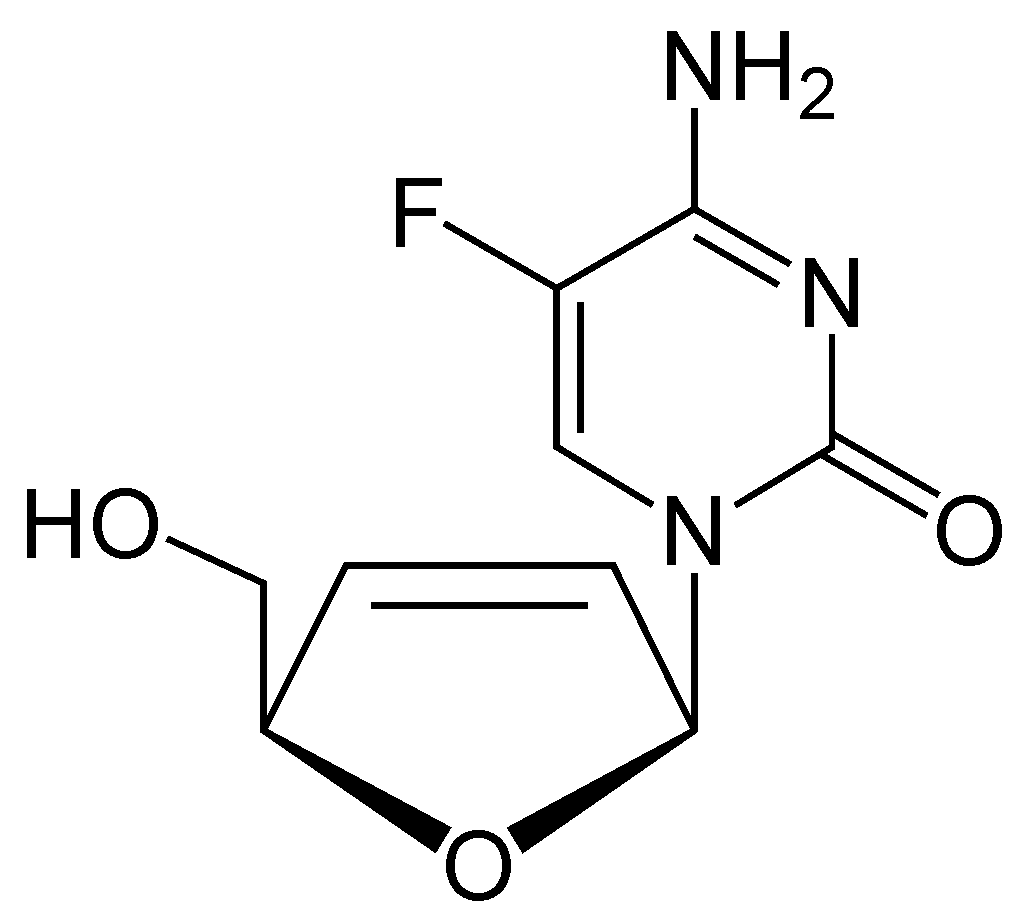
Next in line was 2',3'‐dideoxy‐5-fluoro-3'‐thiacytidine (Emtricitabine, FTC) which is a structural homologue of lamivudine. The structural difference is the 5-fluoro-modification of the base moiety of lamivudine. It is similar in many ways to lamivudine and is active against both HIV-1 and hepatitis B virus (HBV).[21][22]
Carbocyclic nucleoside
Carbocyclic analogues of dideoxyadenosine were investigated for their anti-HIV activity. Minimal activity was first observed. Many nucleoside analogues were prepared and examined but only one had significant activity and satisfied the requirements for clinical use. That was 2´,3´-didehydro analogue of dideoxyadenosine. Insertion of a cyclopropyl group on its 6-amino nitrogen of the adenine ring increased lipophilicity and thus enhanced brain penetration. The resulting compound is known as abacavir (see table 3).[16] Abacavir was approved by the FDA for use in therapy of HIV-1 infections in December 1998.[20]
This drug is the only approved antiretroviral that is active as a guanosine analogue in vivo. First it is monophosphorylated by adenosine phosphotransferase and then the monophosphate is converted to carbovir 3´-monophosphate. Subsequently it is fully phosphorylated and the carbovir is incorporated by the RT into the DNA chain and acts as a chain terminator. Carbovir is a related guanosine analogue that had poor oral bioavailability and thus was withdrawn from clinical development.[19]
Table 3 Comparison of chemical structures: Dideoxyadenosine, didanosine and abacavir Dideoxyadenosine Didanosine Abacavir Chemical structure 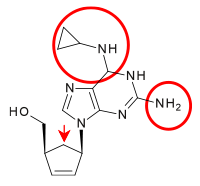
Acyclic nucleotide – the only approved NtRTI
Nucleotide analogues require only two phosphorlylation steps whereas nucleoside analogues require three steps. Reduction[disambiguation needed
] in the phosphorylation requirement may allow more rapid and complete conversion of drugs to their active metabolites. Such considerations have led to the development of phosphonate nucleotide analogues such as tenofovir. Tenofovir disoproxil fumarate (Tenofovir DF) is the prodrug of tenofovir. Tenofovir is an acyclic adenosine derivative. The acyclic nature of the compound and its phosphonate moiety are unique structural features among the approved NRTIs.[21] Tenofovir DF is hydrolyzed enzymatically to tenofovir which exhibits anti-HIV activity.[23][24] It was developed by the synthesis and broad spectrum antiviral activity of 2,3-dihydroxypropyladenine.[24] Tenofovir DF was the first nucleotide reverse-transcriptase inhibitor approved by the FDA for the treatment of HIV-1 infection in October 2001.[18][23]Table 4 A schematic overview of the approved NRTIs and NtRTI and their corresponding endogenous deoxynucleosides and deoxynucleotide. Nucleotide analogue Nucleoside analogues 
Purine analogues
Pyrimidine analogues
Purine analoguesNucleoside 
Adenosine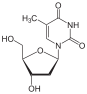
Deoxythymidine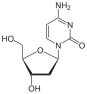
Deoxycytidine
Adenosine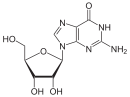
GuanosineDrug 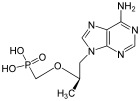
Tenofovir ({[(2R)-1-(6-amino-9H-purin-9-yl)propan-2-yl]oxy}methyl)phosphonic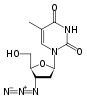
Zidovudine 3´Azido-2´,3´-dideoxythymidine, azidothy midine (AZT)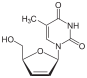
Stavudine 2´,3´-Didehydro-2´,3´-dideoxythymidine (d4T)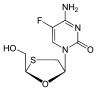
Emtricitabine (-)-ß-L-3´-thia-2´,3´-dideoxy-5-fluorocytidine ( (-)FTC)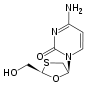
Lamivudine 2´,3´-Dideoxy-3´-thiacytidine (3TC)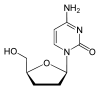
Zalcitabine 2´,3´-Dideoxycycytine (ddC)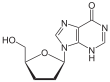
Didanosine 2´,3´-Dideoxyinosine (ddI)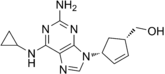
Abacavir (4-(2-amino-6-(cyclopropylamino)-9H-purin-9yl) cyclopent-2enyl)methanol(ABC)Resistance
Currently, appearance of drug resistant viruses is an inevitable consequence of prolonged exposure of HIV-1 to antiretroviral therapy. Drug resistance is a serious clinical concern in treatment of viral infection, and it is a particularly difficult problem in treatment of HIV.[25] Resistance mutations are known for all approved NRTIs.[26]
Two main mechanisms are known that cause NRTI drug resistance: Interference with the incorporation of NRTIs and excision of incorporated NRTIs.[26][27]Interference with the incorporated NRTIs involves a mutation in the p66 subdomain of the RT.[27]The mutation causes a steric hindrance that can exclude certain drugs, for example lamivudine, from being incorporated during reverse transcription. In case of excision of incorporated NRTIs the resistant enzymes readily accept the inhibitor as a substrate for incorporation into the DNA chain.[27] Subsequently the RT enzyme can remove the incorporated NRTI by reversing the polymerization step. The excision reaction requires a pyrophosphate donor which RT joins to the NRTI at the 3´primer terminus, excising it from the primer DNA.[27] To achieve efficient inhibition of HIV-1 replication in patients, and to delay or prevent appearance of drug resistant viruses, drug combinations are used. HAART, also known as highly active antiretroviral therapy consists of combinations of antiviral drugs which include NRTIs, NtRTI, non-nucleoside reverse-transcriptase inhibitors and protease inhibitors.[28]
Current status
Currently, there are several NRTIs in various stages of clinical and preclinical development. The main reasons for continuing the search for new NRTIs against HIV-1 are to decrease toxicity, increase efficiency against resistant viruses, and simplify anti-HIV-1 treatment.[6][26][29]
Apricitabine (ATC)
Apricitabine is a deoxycytidine analogue. It is structurally related to lamivudine where the positions of the oxygen and the sulfur are essentially reversed.[21] Even though apricitabine is a little less potent in vitro compared to some other NRTIs, it maintains its activity against a broad spectrum of HIV-1 variants with NRTI resistance mutations. Apricitabine is in the final stage of clinical development for the treatment of NRTI-experienced patients.[6]
Elvucitabine (L-d4FC)
Elvucitabine is a deoxycytidine analogue with activity against HIV resistant to several other nucleoside analogues, including zidovudine and lamivudine.[22] This is partly because of high intracellular levels of its triphosphate metabolite reached in cells.[6] Clinical trials of elvucitabine are on hold, because it has shown bone marrow suppression in some patients, with CD4+ cell numbers dropping as early as two days after initiation of dosing.[22][29]
Amdoxovir (DAPD)
Amdoxovir is a guanosine analogue NRTI prodrug that has good bioavailability.[6][22][29]It is deaminated intracellularly by adenosine deaminase to dioxolane guanine (DXG). DXG-triphosphate, the active form of the drug, has greater activity than DAPD-triphosphate.[22] Amdoxovir is currently in phasa II clinical trials.[24][29]
Racivir (RCV)
Racivir is a racemic mixture of the two β-enantiomers of emtricitabine (FTC), (-)-FTC and (+)-FTC. Racivir has excellent oral bioavailability and has the advantage of needing to be taken only once a day. Racivir can be considered to be used in combination of two NRTIs and has shown promising antiviral activity when used in combination. Racivir is currently in phase II clinical trials.[6][22][29]
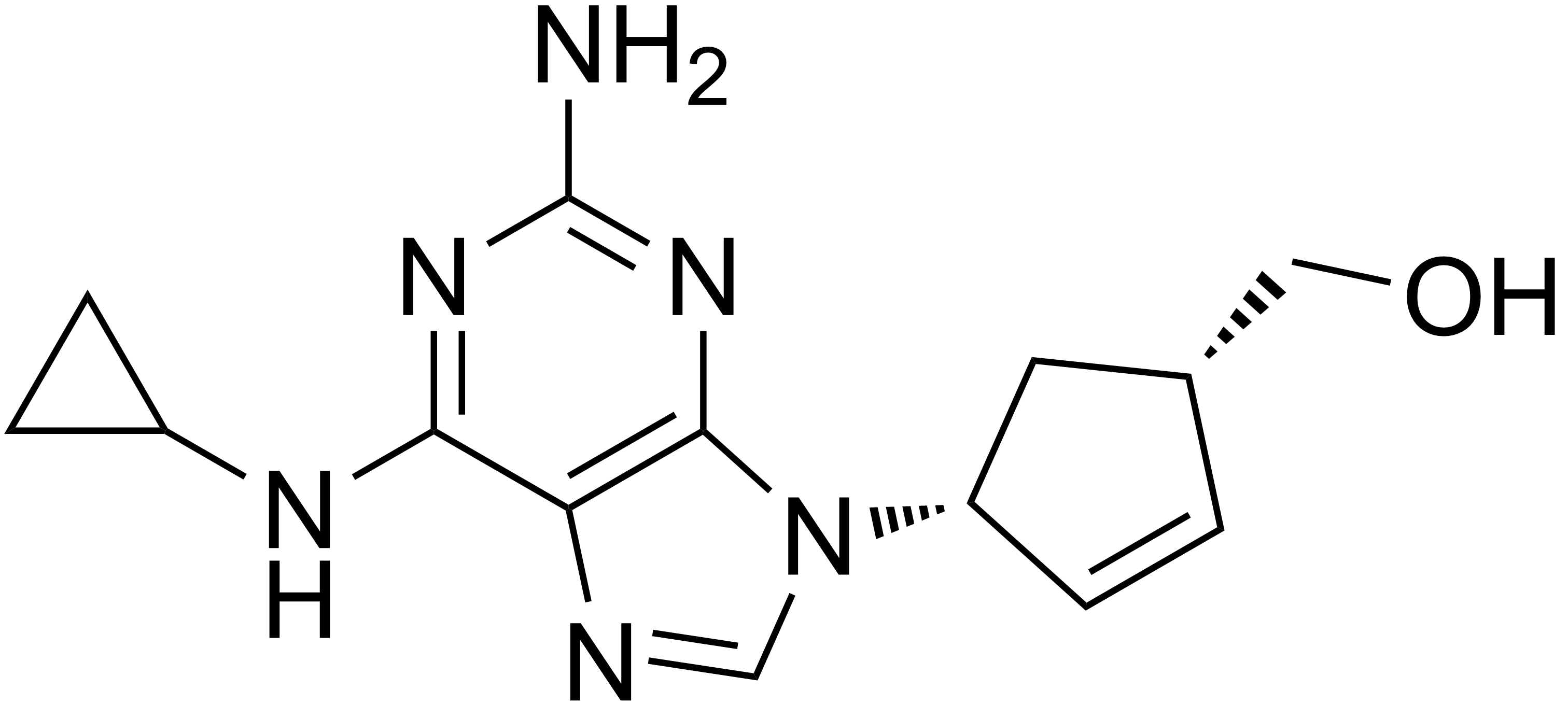
Table 5 Several drug candidates that are undergoing clinical development Drug candidate Apricitabine Elvucitabine Amdoxovir Racivir Chemical structure 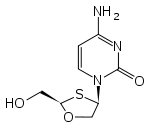
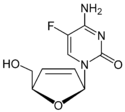
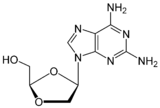
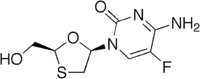
Phase of development Final stage of clinical development On hold phase II phase II There are several more NRTIs in development. Either the sponsors have filed for an Investigational New Drug (IND) application, the application has been approved by the FDA or the drugs are in different phases of clinical trails. Some of the NRTIs that are in development exhibit various attractive pharmacological properties that could make them desirable for the treatment of patients in need of new agents.[6][22][29]
See also
- Antiretroviral drug
- Discovery and development of CCR5 receptor antagonists
- Discovery and Development of Non-Nucleoside Reverse-Transcriptase Inhibitors
- Discovery and Development of HIV Protease Inhibitors
- Reverse-transcriptase inhibitor
- Protease inhibitor
- Entry inhibitor
- Discovery and development of HIV protease inhibitors
- Discovery and development of CCR5 receptor antagonists
- Discovery and development of Bcr-Abl tyrosine kinase inhibitors
References
- ^ Merson, M.D.; Michael, H. (2006), "HIV–AIDS Pandemic at 25 — The Global Response", The New England Journal of Medicine 354 (23): 2414–2417, doi:10.1056/NEJMp068074, http://www.nejm.org/doi/pdf/10.1056/NEJMp068074
- ^ Fausi, A.S. (1999), "The AIDS epidemic Considerations for the 21st Century", The New England Journal of Medicine 351 (14): 1046–1050, doi:10.1056/NEJM199909303411406, http://www.nejm.org/doi/pdf/10.1056/NEJM199909303411406
- ^ a b c Fauci, A.S. (2003), "HIV and AIDS: 20 years of science", Nature medicine 9 (7): 839–843, doi:10.1038/nm0703-839, http://proquest.umi.com/pqdlink?Ver=1&Exp=11-01-2015&FMT=7&DID=1008978971&RQT=309&cfc=1
- ^ a b c De-Clercq, E. (2009), "Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV", International Journal of Antimicrobial Agents 33 (4): 307–320, doi:10.1016/j.ijantimicag.2008.10.010, PMID 19108994, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T7H-4V6RNJX-1-1T&_cdi=5059&_user=3460687&_pii=S0924857908004846&_origin=search&_coverDate=04%2F30%2F2009&_sk=999669995&view=c&wchp=dGLzVtb-zSkWb&md5=3e306038dfc419e3aab9b1fab6412d43&ie=/sdarticle.pdf
- ^ a b Boyer, P.L.; Coffin, J.M.; Delviks_Frankenberry, K.A.; Hughes, S.H.; Jeren, A.; Nikolenko, G.N.; Pathak, V.K. (2008), "HIV-1 reverse transcriptase connection subdomain mutations reduce template RNA degradation and enhance AZT excision", Proceedings of the national academy of sciences of the United States of America 105 (31): 10943–10948, doi:10.1073/pnas.0804660105, http://www.pnas.org/content/105/31/10943.full.pdf+html
- ^ a b c d e f g h i j k l Cihlar, T.; Ray, A.S. (2010), "Nucleoside and nucleotide HIV reverse-transcriptase inhibitors: 25 years after zidovudine", Antiviral research 85 (1): 39–58, doi:10.1016/S0149-2918(00)90004-3, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T2H-4XKHD75-1-F&_cdi=4919&_user=3460687&_pii=S0166354209004859&_origin=search&_coverDate=01%2F31%2F2010&_sk=999149998&view=c&wchp=dGLbVlb-zSkzS&md5=415fb4b14e3ed963994179ac743345ba&ie=/sdarticle.pdf
- ^ a b Herschorn, A.; Hizi (2008), "Retroviral reverse transcriptases (other than those of HIV-1 and murine leukemia virus): A comparison of their molecular and biochemical properties", Virus Research 134: 203–220, doi:10.1016/j.virusres.2007.12.008, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T32-4RYXSHR-1-9&_cdi=4934&_user=3460687&_pii=S0168170207004613&_origin=search&_coverDate=06%2F30%2F2008&_sk=998659998&view=c&wchp=dGLzVzz-zSkWA&md5=3608768b6f39e5560c0c4a5118fdabf4&ie=/sdarticle.pdf
- ^ a b c Giridhar, D.G.; Prajapati; Ramajayam, R.; Yadav, M.R. (2009), "The search for potent, small molecule NNRTIs: A review", Bioorganic & Medicinal Chemistry vol 17: 5744–5762, doi:10.1016/j.bmc.2009.06.060, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6TF8-4WNRJYT-1-31&_cdi=5220&_user=5915660&_orig=search&_coverDate=08%2F15%2F2009&_sk=999829983&view=c&wchp=dGLbVtz-zSkWb&md5=93a1aeea620b65dc46b99a12d4c6790f&ie=/sdarticle.pdf
- ^ a b c Anderson, K.S. (2007), "Reverse transcription of the HIV-1 pandemic", The FASEB Journal 21 (14): 3795–3808, doi:10.1096/fj.07-8697rev, PMID 17639073, http://www.fasebj.org/cgi/reprint/21/14/3795
- ^ a b Goldschmidt, V.; Marquet, R. (2004), "Primer unblocking by HIV-1 reverse transcritptase and resistance to nucleoside RT inhibitors", The international journal of biochemistry and cell biology 36: 1687–1705, doi:10.1016/j.biocel.2004.02.028, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6TCH-4CBVS67-5-D&_cdi=5171&_user=713833&_pii=S1357272504001025&_origin=search&_coverDate=09%2F30%2F2004&_sk=999639990&view=c&wchp=dGLbVzb-zSkWb&md5=cf0c36b9618e1aa54a19699a2aebaa77&ie=/sdarticle.pdf
- ^ Kakuda, T.N. (2010), "Pharmacology of nucleoside and nucleotide Reverse transcriptase inhibitor - induced mitochondrial toxicity", Clinical therapeutics 22 (6): 2717–2747, doi:10.1016/S0149-2918(00)90004-3, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6VRS-416JW04-4-1&_cdi=6242&_user=713833&_pii=S0149291800900043&_origin=search&_coverDate=06%2F30%2F2000&_sk=999779993&view=c&wchp=dGLbVzW-zSkWb&md5=c850f9b8fbe216d0a104a997b998fa3a&ie=/sdarticle.pdf
- ^ Herschorn, A.; Hizi, A. (2010), "Retroviral reverse transcriptases", Cellular and molecular life science 67: 2717–2747, doi:10.1007/s00018-010-0346-2, http://www.springerlink.com/content/l5816755l774w57w/fulltext.pdf
- ^ a b Sneader, W. (1996), Drug prototypes and their exploitation, John Wileys & sons, pp. 448–450, ISBN 0471948470
- ^ a b c William, Hywel (1998), Smith and Williams´ Introduction to the principles of drug design and action, (3 ed.), Harwood academic publishers, pp. 247–250, 486–490, ISBN 90-5702-037-8
- ^ a b Saunders, J. (2000), Top drugs: Top synthetic routes, pp. 71–75
- ^ a b c d e Sneader, W. (2005), Drug discovery a history, pp. 250–268, ISBN 100471899798, http://books.google.is/books?id=mYQxRY9umjcC&pg=PA262&dq=discovery+abacavir&hl=is&ei=PPDOTImiDZKK4gapqtTdDA&sa=X&oi=book_result&ct=result&resnum=8&ved=0CEoQ6AEwBw#v=onepage&q=discovery%20abacavir&f=false
- ^ Georgiev, V.S. (2009), National institute of allergy and infectious diseases, NIH, 2, pp. 417–426, doi:10.1007/978-1-60327-297, ISBN 9781603272694, http://books.google.is/books?id=pymSBkVU-FsC&pg=PA418&dq=discovery+zalcitabine&hl=is&ei=LWHQTLK4LI6P4Aavz72EBw&sa=X&oi=book_result&ct=result&resnum=5&ved=0CDsQ6AEwBDgK#v=onepage&q=discovery%20zalcitabine&f=false
- ^ a b c De-Clercq, E. (2009), "Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV", International Journal of Antimicrobial Agents 33 (4): 307–320, doi:10.1016/j.ijantimicag.2008.10.010, PMID 19108994, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T7H-4V6RNJX-1-1T&_cdi=5059&_user=5915660&_pii=S0924857908004846&_origin=search&_coverDate=04%2F30%2F2009&_sk=999669995&view=c&wchp=dGLbVlz-zSkzV&md5=2cbdd21fbfc59836a9bec8734eae0402&ie=/sdarticle.pdf
- ^ a b Parker, K. (2006), Goodman & Gilman's The Pharmacological Basis of Therapeutics, Eleventh Edition, McGraw-Hill, pp. 1280–1292, ISBN 0-07-142280-3
- ^ a b Ogden, R.C.; Skowron, G. (2006), Reverse transcriptase inhibitors in HIV/AIDS therapy, Humana press inc, pp. 33–63, ISBN 1-58829-649-0
- ^ a b c d LaFemina, R.L. (2009), Antiviral research strategies in antiviral drug discovery, AMS press, pp. 51–70
- ^ a b c d e f g Otto, M.J. (2003), "New nucleoside reverse transcriptase inhibitors for the treatment of HIV infections", Current opinion in pharmacology 9 (7): 839–843, doi:10.1016/j.coph.2004.06.001, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6W7F-4D2FFBK-1-5&_cdi=6625&_user=3460687&_pii=S1471489204001195&_origin=search&_coverDate=10%2F01%2F2004&_sk=999959994&view=c&wchp=dGLbVlW-zSkzV&md5=17a5d272263453f40f9f4f8d660e1ac1&ie=/sdarticle.pdf
- ^ a b Fung, E.A.; Piacenti; Stone (2002), "Tenofovir Disoproxil Fumarate: A Nucleotide ReverseTranscriptase Inhibitor for the Treatment of HIV Infection", Clinical Therapeutics 24 (10): 1515–1548, doi:10.1016/S0149-2918(02)80058-3, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6VRS-47GG8JD-4-1&_cdi=6242&_user=3460687&_pii=S0149291802800583&_origin=search&_coverDate=10%2F31%2F2002&_sk=999759989&view=c&wchp=dGLbVlz-zSkzk&md5=e2e01568d83d543fa47da6b19b0f51d2&ie=/sdarticle.pdf
- ^ a b c Nguyen-Ba, R.F.; Rando (2000), "Development of novel nucleoside analogues for use against drug resistant strains of HIV-1", Drug discovery today 5 (10): 465–476, http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6T64-419C4DN-B-5&_cdi=5020&_user=3460687&_pii=S1359644600015580&_origin=search&_coverDate=10%2F01%2F2000&_sk=999949989&view=c&wchp=dGLbVtb-zSkzk&md5=cac114e05d8bb712dd98b0b98dd90ae0&ie=/sdarticle.pdf
- ^ Arnold, E.; Dasa, K.; Hughesc, S.H.; Lewib, P.J. (2005), "Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors", Progress in Biophysics and Molecular Biology 88: 209–231, http://biomaps.rutgers.edu/DasRTreviewProgBiopMo88_209_2005.pdf
- ^ a b c Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Pathakar, V.K. (2010), "The "Connection" Between HIV Drug Resistance and RNase H", Viruses 2 (7): 1476–1503, doi:10.3390/v2071476, http://www.mdpi.com/1999-4915/2/7/1476/pdf
- ^ a b c d Kirby, K.A.; Marchand, B.; Michailidis, E.; Sarafianos, S.G.; Singh, K. (2010), "Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase", Viruses 2 (2): 606–638, doi:10.3390/v2020606, http://www.mdpi.com/1999-4915/2/2/606/pdf
- ^ Bowling, T.L.; Gu, Z.; L´Heureux, L.; Muys, J.M.; Nguyen-Ba, N.; Rando, R.F.; Wainberg, M.A. (1999), "Mechanism of Action and In Vitro Activity of 1',3'-Dioxolanylpurine Nucleoside Analogues against Sensitive and Drug-Resistant Human Immunodeficiency Virus Type 1 Variants", Antimicrobial Agents and Chemotherapy 43 (10): 2376–2382, http://aac.asm.org/cgi/content/full/43/10/2376
- ^ a b c d e f Agrawala, R.K.; Krishnan, P.N.; Raman, S.; Ravichandran, S.; Veerasamy, R. (2008), "An overview on HIV-1 reverse transcriptase inhibitors", Digest journal of nanomaterials and biostructures 3 (4): 171–187, http://www.chalcogen.infim.ro/Ravichandran-HIV1t.pdf
Drug design steps in design Case studies of discovery and development of drug classes triptans · cephalosporins · antiandrogens · Bcr-Abl tyrosine kinase inhibitors · nucleoside and nucleotide reverse transcriptase inhibitors · melatonin receptor agonists · proton pump inhibitors · angiotensin receptor blockers · dual serotonin and norepinephrine reuptake inhibitors · cyclooxygenase 2 inhibitors · TRPV1 antagonistsCategories:- Nucleoside analog reverse transcriptase inhibitors
- World Health Organization essential medicines
- Drug discovery





















Wikimedia Foundation. 2010.