- Enzyme catalysis
-
Enzyme catalysis is the catalysis of chemical reactions by specialized proteins known as enzymes. Catalysis of biochemical reactions in the cell is vital due to the very low reaction rates of the uncatalysed reactions.
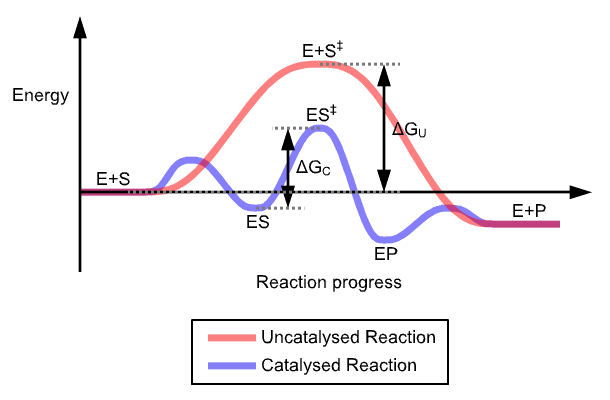
The mechanism of enzyme catalysis is similar in principle to other types of chemical catalysis. By providing an alternative reaction route and by stabilizing intermediates the enzyme reduces the energy required to reach the highest energy transition state of the reaction. The reduction of activation energy (Ea) increases the number of reactant molecules with enough energy to reach the activation energy and form the product.
 Stabilization of the transition state by an enzyme.
Stabilization of the transition state by an enzyme.
Contents
Induced fit
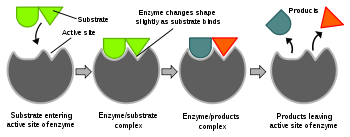 Diagrams to show the induced fit hypothesis of enzyme action.
Diagrams to show the induced fit hypothesis of enzyme action.The favored model for the enzyme-substrate interaction is the induced fit model.[1] This model proposes that the initial interaction between enzyme and substrate is relatively weak, but that these weak interactions rapidly induce conformational changes in the enzyme that strengthen binding.
Catalysis by induced fit
 The different mechanisms of substrate binding
The different mechanisms of substrate bindingThe advantages of the induced fit mechanism arise due to the stabilizing effect of strong enzyme binding. There are two different mechanisms of substrate binding: uniform binding, which has strong substrate binding, and differential binding, which has strong transition state binding. The stabilizing effect of uniform binding increases both substrate and transition state binding affinity, while differential binding increases only transition state binding affinity. Both are used by enzymes and have been evolutionarily chosen to minimize the Ea of the reaction. Enzymes which are saturated, that is, have a high affinity substrate binding, require differential binding to reduce the Ea, whereas small substrate unbound enzymes may use either differential or uniform binding.
These effects have led to most proteins using the differential binding mechanism to reduce the Ea, so most proteins have high affinity of the enzyme to the transition state. Differential binding is carried out by the induced fit mechanism - the substrate first binds weakly, then the enzyme changes conformation increasing the affinity to the transition state and stabilizing it, so reducing the activation energy to reach it.
It is important to clarify, however, that the induced fit concept cannot be used to rationalize catalysis. That is, the chemical catalysis is defined as the reduction of Ea‡ (when the system is already in the ES‡) relative to Ea‡ in the uncatalyzed reaction in water (without the enzyme). The induced fit only suggests that the barrier is lower in the closed form of the enzyme but does not tell us what the reason for the barrier reduction is.
Induced fit may be beneficial to the fidelity of molecular recognition in the presence of competition and noise via the conformational proofreading mechanism .[2]
Mechanisms of transition state stabilization
These conformational changes also bring catalytic residues in the active site close to the chemical bonds in the substrate that will be altered in the reaction. After binding takes place, one or more mechanisms of catalysis lowers the energy of the reaction's transition state, by providing an alternative chemical pathway for the reaction. There are six possible mechanisms of "over the barrier" catalysis as well as a "through the barrier" mechanism:
Catalysis by bond strain
This is the principal effect of induced fit binding, where the affinity of the enzyme to the transition state is greater than to the substrate itself. This induces structural rearrangements which strain substrate bonds into a position closer to the conformation of the transition state, so lowering the energy difference between the substrate and transition state and helping catalyze the reaction.
However, the strain effect is, in fact, a ground state destabilization effect, rather than transition state stabilization effect.[3][4] Furthermore, enzymes are very flexible and they cannot apply large strain effect.[5]
In addition to bond strain in the substrate, bond strain may also be induced within the enzyme itself to activate residues in the active site.
For example: Substrate, bound substrate, and transition state conformations of lysozyme. 
The substrate, on binding, is distorted from the typical 'chair' hexose ring into the 'sofa' conformation, which is similar in shape to the transition state. Catalysis by proximity and orientation
This increases the rate of the reaction as enzyme-substrate interactions align reactive chemical groups and hold them close together. This reduces the entropy of the reactants and thus makes reactions such as ligations or addition reactions more favorable, there is a reduction in the overall loss of entropy when two reactants become a single product.
This effect is analogous to an effective increase in concentration of the reagents. The binding of the reagents to the enzyme gives the reaction intramolecular character, which gives a massive rate increase.
For example: Similar reactions will occur far faster if the reaction is intramolecular. 
The effective concentration of acetate in the intramolecular reaction can be estimated as k2/k1 = 2 x 105 Molar. However, the situation might be more complex, since modern computational studies have established that traditional examples of proximity effects cannot be related directly to enzyme entropic effects.[6][7][8] Also, the original entropic proposal[9] has been found to largely overestimate the contribution of orientation entropy to catalysis.[10]
Catalysis involving proton donors or acceptors (Acid/Base Catalysis)
See also: Protein pKa calculationsProton donors and acceptors, i.e. acids and bases, may donate and accept protons in order to stabilize developing charges in the transition state. This typically has the effect of activating nucleophile and electrophile groups, or stabilizing leaving groups. Histidine is often the residue involved in these acid/base reactions, since it has a pKa close to neutral pH and can therefore both accept and donate protons.
Many reaction mechanisms involving acid/base catalysis assume a substantially altered pKa. This alteration of pKa is possible through the local environment of the residue.
Conditions Acids Bases Hydrophobic environment Increase pKa Decrease pKa Adjacent residues of like charge Increase pKa Decrease pKa Salt bridge (and hydrogen
bond) formationDecrease pKa Increase pKa The pKa is can be modified significantly by the environment, to the extent that residues which are basic in solution may act as proton donors, and vice versa.
For example: Serine protease catalytic mechanism 
The initial step of the serine protease catalytic mechanism involves the histidine of the active site accepting a proton from the serine residue. This prepares the serine as a nucleophile to attack the amide bond of the substrate. This mechanism includes donation of a proton from serine (a base, pKa 14) to histidine (an acid, pKa 6), made possible due to the local environment of the bases. It is important to clarify that the modification of the pKa’s is a pure part of the electrostatic mechanism.[4] Furthermore, the catalytic effect of the above example is mainly associated with the reduction of the pKa of the oxy anion and the increase in the pKa of the histidine, while the proton transfer from the serine to the histidine is not catalyzed significantly, since it is not the rate determining barrier.[11]
Electrostatic catalysis
Stabilization of charged transition states can also be by residues in the active site forming ionic bonds (or partial ionic charge interactions) with the intermediate. These bonds can either come from acidic or basic side chains found on amino acids such as lysine, arginine, aspartic acid or glutamic acid or come from metal cofactors such as zinc. Metal ions are particularly effective and can reduce the pKa of water enough to make it an effective nucleophile.
Systematic computer simulation studies established that electrostatic effects give, by far, the largest contribution to catalysis.[4] In particular, it has been found that enzyme provides an environment which is more polar than water, and that the ionic transition states are stabilized by fixed dipoles. This is very different from transition state stabilization in water, where the water molecules must pay with "reorganization energy".[12] in order to stabilize ionic and charged states. Thus, the catalysis is associated with the fact that the enzyme polar groups are preorganized [13]
For example: Carboxypeptidase catalytic mechanism 
The tetrahedral intermediate is stabilised by a partial ionic bond between the Zn2+ ion and the negative charge on the oxygen. Covalent catalysis
Covalent catalysis involves the substrate forming a transient covalent bond with residues in the active site or with a cofactor. This adds an additional covalent intermediate to the reaction, and helps to reduce the energy of later transition states of the reaction. The covalent bond must, at a later stage in the reaction, be broken to regenerate the enzyme. This mechanism is found in enzymes such as proteases like chymotrypsin and trypsin, where an acyl-enzyme intermediate is formed. Schiff base formation using the free amine from a lysine residue is another mechanism, as seen in the enzyme aldolase during glycolysis.
Some enzymes utilize non-amino acid cofactors such as pyridoxal phosphate (PLP) or thiamine pyrophosphate (TPP) to form covalent intermediates with reactant molecules.[14][15] Such covalent intermediates function to reduce the energy of later transition states, similar to how covalent intermediates formed with active site amino acid residues allow stabilization, but the capabilities of cofactors allow enzymes to carryout reactions that amino acid side residues alone could not. Enzymes utilizing such cofactors include the PLP-dependent enzyme aspartate transaminase and the TPP-dependent enzyme pyruvate dehydrogenase.[16][17]
It is important to clarify that covalent catalysis does correspond in most cases to simply the use of a specific mechanism rather than to true catalysis.[4] For example, the energetics of the covalent bond to the serine molecule in chymotrypsin should be compared to the well-understood covalent bond to the nucleophile in the uncatalyzed solution reaction. A true proposal of a covalent catalysis (where the barrier is lower than the corresponding barrier in solution) would require, for example, a partial covalent bond to the transition state by an enzyme group (e.g., a very strong hydrogen bond), and such effects do not contribute significantly to catalysis.
Quantum tunneling
These traditional "over the barrier" mechanisms have been challenged in some cases by models and observations of "through the barrier" mechanisms (quantum tunneling). Some enzymes operate with kinetics which are faster than what would be predicted by the classical ΔG‡. In "through the barrier" models, a proton or an electron can tunnel through activation barriers.[18][19] Quantum tunneling for protons has been observed in tryptamine oxidation by aromatic amine dehydrogenase.[20]
Interestingly, quantum tunneling does not appear to provide a major catalytic advantage, since the tunneling contributions are similar in the catalyzed and the uncatalyzed reactions in solution.[19][21][22][23] However, the tunneling contribution (typically enhancing rate constants by a factor of ~1000[20] compared to the rate of reaction for the classical 'over the barrier' route) is likely crucial to the viability of biological organisms. This emphasizes the general importance of tunneling reactions in biology.
In 1971-1972 the first quantum-mechanical model of enzyme catalysis was formulated.[24][25]
Examples of catalytic mechanisms
In reality, most enzyme mechanisms involve a combination of several different types of catalysis.
Triose phosphate isomerase
Triose phosphate isomerase (EC 5.3.1.1) catalyses the reversible interconvertion of the two triose phosphates isomers dihydroxyacetone phosphate and D-glyceraldehyde 3-phosphate.
Trypsin
Trypsin (EC 3.4.21.4) is a serine protease that cleaves protein substrates at lysine and arginine amino acid residues.
Aldolase
Aldolase (EC 4.1.2.13) catalyses the breakdown of fructose 1,6-bisphosphate (F-1,6-BP) into glyceraldehyde 3-phosphate and dihydroxyacetone phosphate (DHAP).
See also
- Enzyme assay
- Enzyme kinetics
- Protein dynamics
- Quantum tunnelling
- The Proteolysis Map
- Time resolved crystallography
References
- ^ Koshland DE (February 1958). "Application of a Theory of Enzyme Specificity to Protein Synthesis". Proc. Natl. Acad. Sci. U.S.A. 44 (2): 98–104. doi:10.1073/pnas.44.2.98. PMC 335371. PMID 16590179. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=335371.
- ^ Savir Y & Tlusty T (2007). Scalas, Enrico. ed. "Conformational Proofreading: The Impact of Conformational Changes on the Specificity of Molecular Recognition". PLoS ONE 2 (5): e468. doi:10.1371/journal.pone.0000468. PMC 1868595. PMID 17520027. http://www.weizmann.ac.il/complex/tlusty/papers/PLoSONE2007.pdf.
- ^ Jencks W.P. "Catalysis in Chemistry and Enzymology" 1987, Dover, New York
- ^ a b c d Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H.M. (2006). "Electrostatic Basis of Enzyme Catalysis". Chem. Rev. 106 (8): 3210–3235. doi:10.1021/cr0503106. PMID 16895325.
- ^ Warshel, A.; Levitt, M. (1976). "Theoretical Studies of Enzymatic Reactions: Dielectric Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme". J. Mol. Biol. 103 (2): 227–49. doi:10.1016/0022-2836(76)90311-9. PMID 985660.
- ^ Stanton, R.V.; Perakyla, M.; Bakowies, D.; Kollman, P.A. (1998). "Combined ab initio and Free Energy Calculations To Study Reactions in Enzymes and Solution: Amide Hydrolysis in Trypsin and Aqueous Solution". J. Am. Chem. Soc 120 (14): 3448–3457. doi:10.1021/ja972723x.
- ^ Kuhn, B.; Kollman, P.A. (2000). "QM-FE and Molecular Dynamics Calculations on Catechol O-Methyltransferase: Free Energy of Activation in the Enzyme and in Aqueous Solution and Regioselectivity of the Enzyme-Catalyzed Reaction". J. Am. Chem. Soc 122 (11): 2586–2596. doi:10.1021/ja992218v.
- ^ Bruice, T.C.; Lightstone, F.C. (1999). "Ground State and Transition State Contributions to the Rates of Intramolecular and Enzymatic Reactions". Acc. Chem. Res. 32 (2): 127–136. doi:10.1021/ar960131y.
- ^ Page, M.I.; Jencks, W.P.. "Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect". Proc. Natl. Acad. Sci. USA 1971 (68): 1678–1683. PMC 389269. PMID 5288752. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=389269.
- ^ Warshel, A.; Parson, W.W. (2001). "Dynamics of Biochemical and Biophysical Reactions: Insight from Computer Simulations". Quart. Rev. Biophys 34: 563–679.
- ^ Warshel, A.; Naray-Szabo, G.; Sussman, F.; Hwang, J.-K. (1989). "How do Serine Proteases Really Work?". Biochemistry 1989 (28): 3629–37. doi:10.1021/bi00435a001. PMID 2665806.
- ^ Marcus R. A. "On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions" J. Chem. Phys. 1965 43:679-701
- ^ Warshel A. "Energetics of Enzyme Catalysis", Proc. Natl. Acad. Sci. USA, 1978 75: 5250
- ^ Toney, M. D. "Reaction specificity in pyridoxal enzymes." Archives of biochemistry and biophysics (2005) 433: 279-287
- ^ Micronutrient Information Center, Oregon State University
- ^ Voet, Donald; Judith Voet (2004). Biochemistry. John Wiley & Sons Inc. pp. 986–989. ISBN 0-471-25090-2.
- ^ Voet, Donald; Judith Voet (2004). Biochemistry. John Wiley & Sons Inc. pp. 604–606. ISBN 0-471-25090-2.
- ^ Garcia-Viloca, M; Gao, J; Karplus, M; Truhlar, DG (2004). "How enzymes work: analysis by modern rate theory and computer simulations". Science 303 (5655): 186–95. doi:10.1126/science.1088172. PMID 14716003.
- ^ a b Olsson, MH; Siegbahn, PE; Warshel, A (2004). "Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase". Journal of the American Chemical Society 126 (9): 2820–8. doi:10.1021/ja037233l. PMID 14995199.
- ^ a b Masgrau, L; Roujeinikova, A; Johannissen, LO; Hothi, P; Basran, J; Ranaghan, KE; Mulholland, AJ; Sutcliffe, MJ et al. (2006). "Atomic description of an enzyme reaction dominated by proton tunneling". Science 312 (5771): 237–41. doi:10.1126/science.1126002. PMID 16614214.
- ^ Hwang, J.-K.; Warshel, A.. "How important are quantum mechanical nuclear motions in enzyme catalysis". J. Am. Chem. Soc 1996 (118): 11745–11751.
- ^ Ball, P. (2004). "Enzymes: By chance, or by design?". Nature 431 (7007): 396. Bibcode 2004Natur.431..396B. doi:10.1038/431396a.
- ^ Olsson, M.H.M.; Parson, W.W.; Warshel, A.. "Dynamical Contributions to Enzyme Catalysis: Critical Tests of A Popular Hypothesis". Chem. Rev. 2006 (105): 1737–1756.
- ^ Volkenshtein M.V., Dogonadze R.R., Madumarov A.K., Urushadze Z.D., Kharkats Yu.I. Theory of Enzyme Catalysis.- Molekuliarnaya Biologia, Moscow, 6, 1972, 431-439
- ^ Volkenshtein M.V., Dogonadze R.R., Madumarov A.K., Urushadze Z.D., Kharkats Yu.I. Electronic and Conformational Interactions in Enzyme Catalysis. In: E.L. Andronikashvili (Ed.), Konformatsionnie Izmenenia Biopolimerov v Rastvorakh, Publishing House "Nauka", Moscow, 1973, 153-157
Further reading
- Alan Fersht, Structure and Mechanism in Protein Science : A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, 1998. ISBN 0-7167-3268-8
- Dedicated issue of Philosophical Transactions B on Quantum catalysis in enzymes freely available.
Concepts in asymmetric synthesis Chirality types Chirality · Stereocenter · Planar chirality · Chiral ligand · Axial chirality · Supramolecular chirality · Inherent chiralityChiral molecules Stereoisomer · Enantiomer · Diastereomer · Meso compound · Enantiomeric excess · Diastereomeric excess ·Analysis Optical rotation · Chiral derivatizing agents · NMR spectroscopy of stereoisomers · Ultraviolet-visible spectroscopy of stereoisomersChiral resolution Recrystallization · Kinetic resolution · Chiral column chromatography · Diastereomeric recrystallizationReactions Proteins: enzymes Topics Active site · Allosteric regulation · Binding site · Catalytically perfect enzyme · Coenzyme · Cofactor · Cooperativity · EC number · Enzyme catalysis · Enzyme inhibitor · Enzyme kinetics · Lineweaver–Burk plot · Michaelis–Menten kinetics · List of enzymesTypes EC1 Oxidoreductases/list · EC2 Transferases/list · EC3 Hydrolases/list · EC4 Lyases/list · EC5 Isomerases/list · EC6 Ligases/listB enzm: 1.1/2/3/4/5/6/7/8/10/11/13/14/15-18, 2.1/2/3/4/5/6/7/8, 2.7.10, 2.7.11-12, 3.1/2/3/4/5/6/7, 3.1.3.48, 3.4.21/22/23/24, 4.1/2/3/4/5/6, 5.1/2/3/4/99, 6.1-3/4/5-6 Categories:- Enzymes
- Catalysis


Wikimedia Foundation. 2010.