- Ellis–van Creveld syndrome
-
Ellis–van Creveld syndrome Classification and external resources 
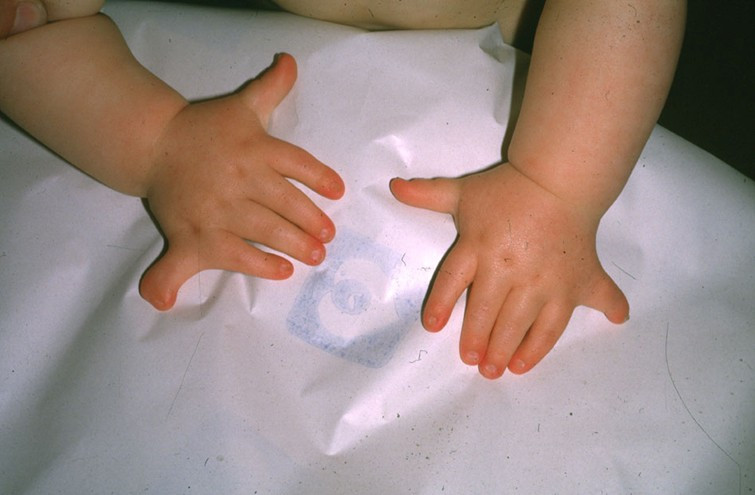
Polydactyly in Ellis–van Creveld syndromeICD-10 Q77.6 ICD-9 756.55 OMIM 225500 DiseasesDB 29309 MedlinePlus 001667 eMedicine ped/660 MeSH D004613 Ellis–van Creveld Syndrome (also called chondroectodermal dysplasia or mesoectodermal dysplasia) is a rare genetic disorder of the skeletal dysplasia type.
Contents
Symptoms
It involves numerous anomalies including post-axial polydactyly, congenital heart defects (most commonly an atrial septal defect producing a common atrium, occurring in 60% of affected individuals), pre-natal tooth eruption, fingernail dysplasia, short-limbed dwarfism, short ribs, cleft palate, and malformation of the wrist bones (fusion of the hamate and capitate bones).
Genetics
 Ellis–van Creveld syndrome has an autosomal recessive pattern of inheritance.
Ellis–van Creveld syndrome has an autosomal recessive pattern of inheritance.
Ellis–van Creveld syndrome often is the result of founder effects in isolated human populations, such as the Amish and some small island inhabitants. Although relatively rare, this disorder does occur with higher incidence within founder-effect populations due to lack of genetic variability. Observation of the inheritance pattern has illustrated that the disease is autosomal recessive, meaning that both parents have to carry the gene in order for an individual to be affected by the disorder.[1]
Ellis–van Creveld syndrome is caused by a mutation in the EVC gene, as well as by a mutation in a nonhomologous gene, EVC2, located close to the EVC gene in a head-to-head configuration. The gene was identified by positional cloning.[2] The EVC gene maps to the chromosome 4 short arm. The function of a healthy EVC gene is not well understood at this time.[1]
Relation to other rare disorders: genetic ciliopathy
Until recently, the medical literature did not indicate a connection among many genetic disorders, both genetic syndromes and genetic diseases, that are now being found to be related. As a result of new genetic research, some of these are, in fact, highly related in their root cause despite the widely-varying set of medical symptoms that are clinically visible in the disorders. Ellis–van Creveld syndrome is one such disease, part of an emerging class of diseases called ciliopathies. The underlying cause may be a dysfunctional molecular mechanism in the primary cilia structures of the cell, organelles which are present in many cellular types throughout the human body. The cilia defects adversely affect "numerous critical developmental signaling pathways" essential to cellular development and thus offer a plausible hypothesis for the often multi-symptom nature of a large set of syndromes and diseases. Known ciliopathies include primary ciliary dyskinesia, Bardet–Biedl syndrome, polycystic kidney and liver disease, nephronophthisis, Alstrom syndrome, Meckel–Gruber syndrome and some forms of retinal degeneration.[3]
History
The disorder was described by Richard W. B. Ellis (1902–1966) of Edinburgh and Simon van Creveld (1895–1971) of Amsterdam.[4] Each had a patient with this syndrome, as they had discovered when they met in the same train compartment on the way to a pediatrics conference in England in the late 1930s. A third patient had been referred to by L. Emmett Holt, Jr. and Rustin McIntosh in a textbook of pediatrics (Holt and McIntosh, 1933) and was included in full in the paper by Ellis and van Creveld (1940).[5]
McCusick et al.(1964) followed up with a study of its incidence in the Amish population. He observed the largest pedigree so far, in an inbred religious isolate, the Old Order Amish, in Lancaster County, Pennsylvania.[6] Almost as many persons were known in this one kindred as had been reported in all the medical literature up to that time.
Nomenclature
'Six-fingered dwarfism' ('digital integer deficiency') was an alternative designation used for this condition when it was being studied in the Amish[6] and may have served a useful function in defining this then little known condition for the medical profession, as well as the lay public. The term, however, has been found offensive by some, apparently not because of 'dwarfism,' but because of the reference to the polydactyly, which is seen as a 'freakish' labeling. For this reason, 6-fingered dwarfism has been removed as an alternative name for this entry. This leaves Ellis–van Creveld syndrome with its felicitous abbreviation, EVC, as the only satisfactory designation. Chondroectodermal dysplasia and mesoectodermal dysplasia do not well define the entity and are not satisfactory for general usage, either medical or otherwise.
References
- ^ a b "Ellis-van Creveld syndrome". Genes and Diseases. NCBI. 1998. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gnd&part=ellisvancreveldsyndrome. Retrieved November 8, 2010.
- ^ Ruiz-Perez VL, Ide SE, Strom TM, et al. (2000). "Mutations in a new gene in Ellis–van Creveld syndrome and Weyers acrodental dysostosis". Nat. Genet. 24 (3): 283–6. doi:10.1038/73508. PMID 10700184.
- ^ Badano, Jose L.; Norimasa Mitsuma, Phil L. Beales, Nicholas Katsanis (September 2006). "The Ciliopathies : An Emerging Class of Human Genetic Disorders". Annual Review of Genomics and Human Genetics 7: 125–148. doi:10.1146/annurev.genom.7.080505.115610. PMID 16722803. http://arjournals.annualreviews.org/doi/abs/10.1146/annurev.genom.7.080505.115610. Retrieved 2008-06-15.
- ^ synd/1114 at Who Named It?
- ^ Ellis, R. W. B.; van Creveld, S.: A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: report of three cases. Arch. Dis. Child. 15: 65–84, 1940.
- ^ a b McKusick, V. A.; Egeland, J. A.; Eldridge, R.; Krusen, D. E. (1964). "Dwarfism in the Amish. I. The Ellis–van Creveld syndrome". Bulletin of the Johns Hopkins Hospital 115: 306–36. PMID 14217223.
External links
Osteochondrodysplasia (Q77–Q78, 756.4–756.5) Osteodysplasia/
osteodystrophyOther/ungroupedFLNB (Boomerang dysplasia) · Opsismodysplasia · Polyostotic fibrous dysplasia (McCune-Albright syndrome)Chondrodysplasia/
chondrodystrophy
(including dwarfism)enchondromatosis (Ollier disease, Maffucci syndrome)Other dwarfismFibrochondrogenesis · Short rib-polydactyly syndrome (Majewski's polydactyly syndrome) · Léri-Weill dyschondrosteosisCongenital malformations and deformations of integument / skin disease (Q80–Q82, 757.0–757.3) Genodermatosis Congenital ichthyosis/
erythrokeratodermiaADARUngroupedIchthyosis bullosa of Siemens · Ichthyosis follicularis · Ichthyosis prematurity syndrome · Ichthyosis–sclerosing cholangitis syndrome · Nonbullous congenital ichthyosiform erythroderma · Ichthyosis linearis circumflexa · Ichthyosis hystrixEB
and relatedJEB (JEB-H, Mitis, Generalized atrophic, JEB-PA)related: Costello syndrome · Kindler syndrome · Laryngoonychocutaneous syndrome · Skin fragility syndrome ·Naegeli syndrome/Dermatopathia pigmentosa reticularis · Hay–Wells syndrome · Hypohidrotic ectodermal dysplasia · Focal dermal hypoplasia · Ellis–van Creveld syndrome · Rapp–Hodgkin syndrome/Hay–Wells syndromeEhlers–Danlos syndrome · Cutis laxa (Gerodermia osteodysplastica) · Popliteal pterygium syndrome · Pseudoxanthoma elasticum · Van Der Woude syndromeHyperkeratosis/
keratinopathydiffuse: Diffuse epidermolytic palmoplantar keratoderma • Diffuse nonepidermolytic palmoplantar keratoderma • Palmoplantar keratoderma of Sybert • Mal de Meleda •syndromic (connexin (Bart–Pumphrey syndrome • Clouston's hidrotic ectodermal dysplasia • Vohwinkel syndrome) • Corneodermatoosseous syndrome • plakoglobin (Naxos syndrome) • Scleroatrophic syndrome of Huriez • Olmsted syndrome • Cathepsin C (Papillon–Lefèvre syndrome • Haim–Munk syndrome) • Camisa diseasefocal: Focal palmoplantar keratoderma with oral mucosal hyperkeratosis • Focal palmoplantar and gingival keratosis • Howel–Evans syndrome • Pachyonychia congenita (Pachyonychia congenita type I • Pachyonychia congenita type II) • Striate palmoplantar keratoderma • Tyrosinemia type II)punctate: Acrokeratoelastoidosis of Costa • Focal acral hyperkeratosis • Keratosis punctata palmaris et plantaris • Keratosis punctata of the palmar creases • Schöpf–Schulz–Passarge syndrome • Porokeratosis plantaris discreta • Spiny keratodermaungrouped: Palmoplantar keratoderma and spastic paraplegia • desmoplakin (Carvajal syndrome) • connexin (Erythrokeratodermia variabilis • HID/KID)OtherMeleda disease · Keratosis pilaris · ATP2A2 (Darier's disease) · Dyskeratosis congenita · Lelis syndromeDyskeratosis congenita · Keratolytic winter erythema · Keratosis follicularis spinulosa decalvans · Keratosis linearis with ichthyosis congenital and sclerosing keratoderma syndrome · Keratosis pilaris atrophicans faciei · Keratosis pilarisOthercadherin (EEM syndrome) · immune system (Hereditary lymphedema, Mastocytosis/Urticaria pigmentosa) · Hailey–Hailey
see also Template:Congenital malformations and deformations of skin appendages, Template:Phakomatoses, Template:Pigmentation disorders, Template:DNA replication and repair-deficiency disorderDevelopmental
anomaliesMidlineOther/ungroupedAplasia cutis congenita · Amniotic band syndrome · Branchial cyst · Cavernous venous malformation
Accessory nail of the fifth toe · Bronchogenic cyst · Congenital cartilaginous rest of the neck · Congenital hypertrophy of the lateral fold of the hallux · Congenital lip pit · Congenital malformations of the dermatoglyphs · Congenital preauricular fistula · Congenital smooth muscle hamartoma · Cystic lymphatic malformation · Median raphe cyst · Melanotic neuroectodermal tumor of infancy · Mongolian spot · Nasolacrimal duct cyst · Omphalomesenteric duct cyst · Poland anomaly · Rapidly involuting congenital hemangioma · Rosenthal–Kloepfer syndrome · Skin dimple · Superficial lymphatic malformation · Thyroglossal duct cyst · Verrucous vascular malformation · BirthmarkCategories:- Autosomal recessive disorders
- Genodermatoses
- Syndromes

Wikimedia Foundation. 2010.