- Hexosaminidase
-
β-N-acetylhexosaminidase 
Hexosaminidase A (Hex A) Identifiers EC number 3.2.1.52 CAS number 9012-33-3 Databases IntEnz IntEnz view BRENDA BRENDA entry ExPASy NiceZyme view KEGG KEGG entry MetaCyc metabolic pathway PRIAM profile PDB structures RCSB PDB PDBe PDBsum Gene Ontology AmiGO / EGO Search PMC articles PubMed articles Hexosaminidase is an enzyme involved in the hydrolysis of terminal N-acetyl-D-hexosamine residues in N-acetyl-β-D-hexosaminides.
Contents
Isozymes and genes
Lysosomal A, B, and S isozymes
Functional lysosomal β-hexosaminidase enzymes are dimeric in structure. Three isozymes are produced through the combination of α and β subunits to form any one of three active dimers:[1]
hexosaminidase
isozymesubunit composition function A α/β heterodimer only isozyme that can hydrolyze GM2 ganglioside in vivo B β/β homodimer exists in tissues but no known physiological function S α/α homodimer exists in tissues but no known physiological function The α and β subunits are encoded by separate genes, HEXA and HEXB respectively. Beta-hexosaminidase and the cofactor GM2 activator protein catalyze the degradation of the GM2 gangliosides and other molecules containing terminal N-acetyl hexosamines.[2] Gene mutations in HEXB often result in Sandhoff disease; whereas, mutations in HEXA decrease the hydrolysis of GM2 gangliosides, which is the main cause of Tay-Sachs disease.[3]
β-hexosaminidase subunit alpha Identifiers Symbol HEXA Entrez 3073 HUGO 4878 OMIM 606869 RefSeq NM_000520 UniProt P06865 Other data EC number 3.2.1.52 Locus Chr. 15 q24.1 β-hexosaminidase subunit beta Identifiers Symbol HEXB Entrez 3074 HUGO 4879 OMIM 606873 RefSeq NM_000521 UniProt P07686 Other data EC number 3.2.1.52 Locus Chr. 5 q13.3 Function
Even though the alpha and beta subunits of lysosomal hexosaminidase can both cleave GalNAc residues, only the alpha subunit is able to hydrolyze GM2 gangliosides because of a key residue, Arg-424, and a loop structure that forms from the amino acid sequence in the alpha subunit. The loop in the alpha subunit , consisting of Gly-280, Ser-281, Glu-282, and Pro-283 which is absent in the beta subunit, serves as an ideal structure for the binding of the GM2 activator protein (GM2AP), and arginine is essential for binding the N-acetyl-neuraminic acid residue of GM2 gangliosides. The GM2 activator protein transports GM2 gangliosides and presents the lipids to hexosaminidase, so a functional hexosaminidase enzyme is able to hydrolyze GM2 gangliosides into GM3 gangliosides by removing the N-acetylgalactosamine (GalNAc) residue from GM2 gangliosides.[4]
Mechanism of action
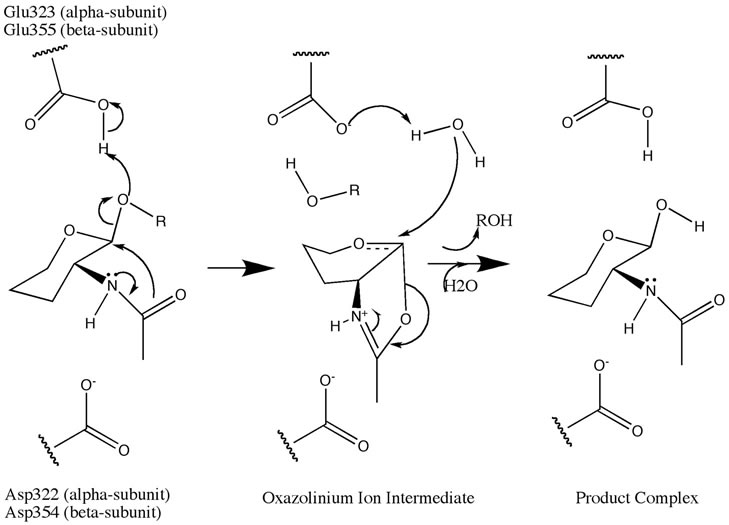
A Michaelis complex consisting of a glutamate residue, a GalNAc residue on the GM2 ganglioside, and an aspartate residue leads to the formation of an oxazolinium ion intermediate. A glutamate residue (alpha Glu-323/beta Glu-355) works as an acid by donating its hydrogen to the glycosidic oxygen atom on the GalNAc residue. An aspartate residue (alpha Asp-322/beta Asp-354) positions the C2-acetamindo group so that it can be attacked by the nucleophile (N-acetamido oxygen atom on carbon 1 of the substrate). The aspartate residue stabilizes the positive charge on the nitrogen atom in the oxazolinium ion intermediate. Following the formation of the oxazolinium ion intermediate, water attacks the electrophillic acetal carbon. Glutamate acts as a base by deprotonating the water leading to the formation of the product complex and the GM3ganglioside.[4]
Gene mutations resulting in Tay-Sachs Disease
There are numerous mutations that lead to hexosaminidase deficiency including gene deletions, nonsense mutations, and missense mutations. Tay-Sachs disease occurs when hexosaminidase A loses its ability to function. People with Tay-Sachs disease are unable to remove the GalNAc residue from the GM2 ganglioside, and as a result, they end up storing 100 to 1000 times more GM2 gangliosides in the brain than the normal person. Over 100 different mutations have been discovered just in infantile cases of Tay-Sachs disease alone.[5]
The most common mutation, which occurs in over 80 percent of Tay-Sachs patients, results from a four base pair addition (TATC) in exon 11 of the Hex A gene. This insertion leads to an early stop codon, which causes the Hex A deficiency.[6]
Children born with Tay-Sachs usually die between two to four years of age from aspiration and pneumonia. Tay-Sachs causes cerebral degeneration and blindness. Patients also experience flaccid extremities and seizures. At this point in time, there has been no cure or effective treatment of Tay-Sachs disease.[5]
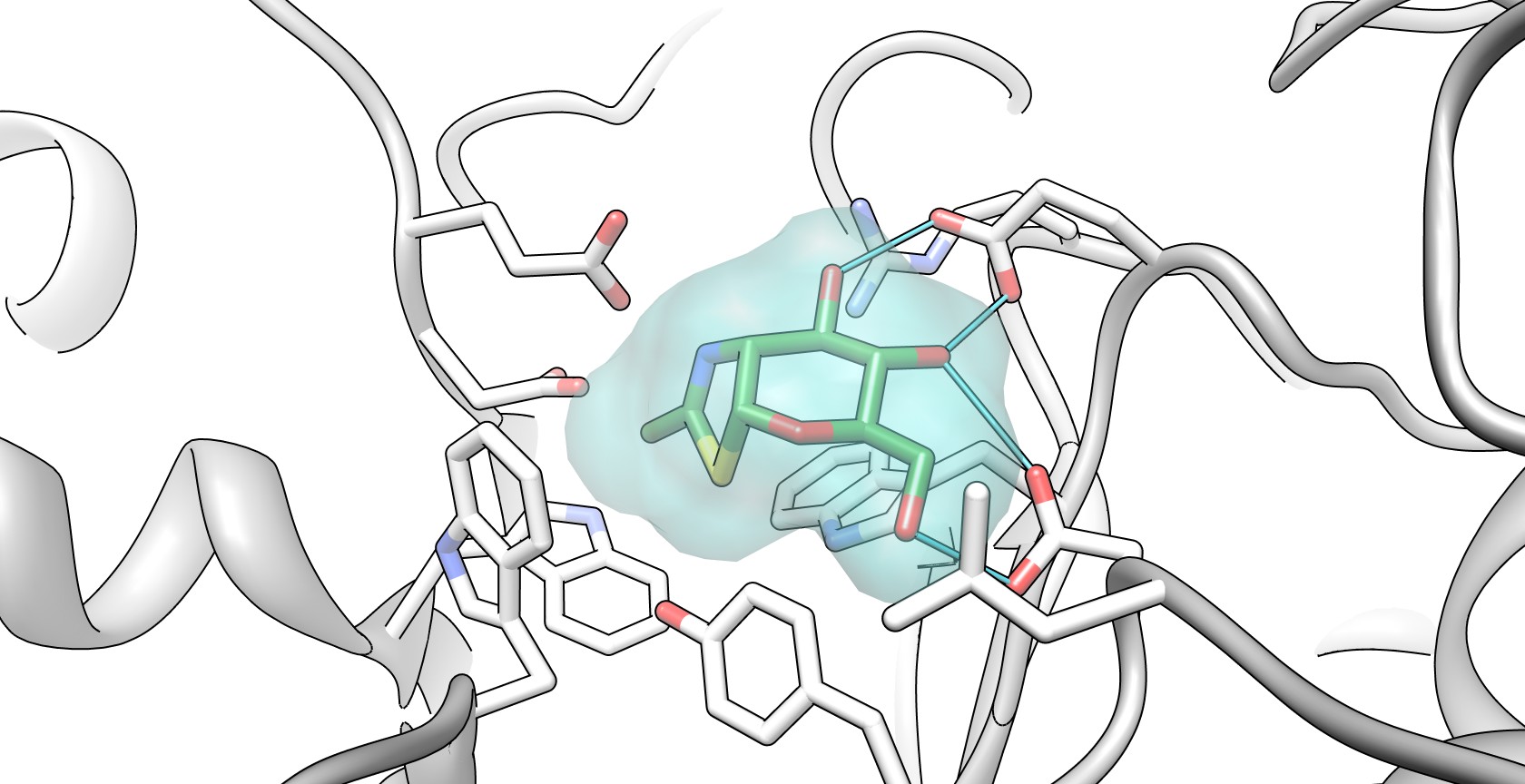
NAG-thiazoline, NGT, acts as mechanism based inhibitor of hexosaminidase A. In patients with Tay-Sachs disease (misfolded hexosaminidase A), NGT acts as a molecular chaperone by binding in the active site of hexosaminidase A which helps create a properly folded hexosaminidase A. The stable dimer conformation of hexosaminidase A has the ability to leave the endoplasmic reticulum and is directed to the lysosome where it can perform the degradation of GM2 gangliosides.[4] The two subunits of hexosaminidase A are shown below:
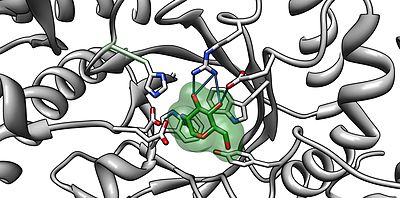 The alpha subunit active site shown bound to NAG-thiazoline (NGT) in beta-hexosaminidase. PDB 2GK1 The light green outline surrounding NGT represents the Van der Waals surface of NGT. The critical amino acids in the active site that are able to hydrogen bond with NGT include Arginine 178 and Glutamate 462.[4]
The alpha subunit active site shown bound to NAG-thiazoline (NGT) in beta-hexosaminidase. PDB 2GK1 The light green outline surrounding NGT represents the Van der Waals surface of NGT. The critical amino acids in the active site that are able to hydrogen bond with NGT include Arginine 178 and Glutamate 462.[4]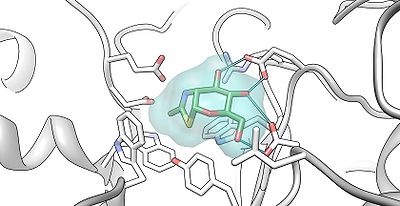 The beta subunit active site shown bound to NAG-thiazoline (NGT) in beta-hexosaminidase. PDB 2GK1 The light blue outline surrounding NGT represents the Van der Waals surface of NGT. The critical amino acids in the active site that are able to hydrogen bond with NGT include Glutamate 491 and Aspartate 452.[4]
The beta subunit active site shown bound to NAG-thiazoline (NGT) in beta-hexosaminidase. PDB 2GK1 The light blue outline surrounding NGT represents the Van der Waals surface of NGT. The critical amino acids in the active site that are able to hydrogen bond with NGT include Glutamate 491 and Aspartate 452.[4]Cytosolic C and D isozymes
The bifunctional protein NCOAT (nuclear cytoplasmic O-GlcNAcase and acetyltransferase) that is encoded by the MGEA5 gene possesses both hexosaminidase and histone acetyltransferase activities.[7] NCOAT is also known as hexosaminidase C[8] and has distinct substrate specificities compared to lysosomal hexosaminidase A.[9] A single-nucleotide polymorphism in the human O-GlcNAcase gene is linked to diabetes mellitus type 2.[10]
A fourth mammalian hexosaminidase polypeptide which has been designated hexosaminidase D (HEXDC) has recently been identified.[11]
hexosaminidase C Identifiers Symbol MGEA5 Entrez 10724 HUGO 7056 OMIM 604039 RefSeq NM_012215 UniProt O60502 Other data EC number 3.2.1.52 Locus Chr. 10 q24.1-24.3 hexosaminidase D Identifiers Symbol HEXDC Alt. symbols FLJ23825 Entrez 284004 HUGO 26307 RefSeq NM_173620 UniProt Q8IYN4 Other data EC number 3.2.1.52 Locus Chr. 17 q25.3 References
- ^ Hou Y, Tse R, Mahuran DJ (April 1996). "Direct determination of the substrate specificity of the alpha-active site in heterodimeric beta-hexosaminidase". Biochemistry 35 (13): 3963–9. doi:10.1021/bi9524575. PMID 8672428.
- ^ Knapp S, Vocadlo D, Gao Z, Kirk B, Lou J, Withers SG (1996). "NAG-thiazoline, an N-acetylbeta-hexosaminidase inhibitor that implicates acetamido participation". J. Am. Chem. Soc. 118 (28): 6804–6805. doi:10.1021/ja960826u.
- ^ Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN (April 2003). "Crystal structure of human beta-hexosaminidase: understanding the molecular basis of Sandhoff and Tay-Sachs disease". J. Mol. Biol. 327 (5): 1093–109. doi:10.1016/S0022-2836(03)00216-X. PMC 2910754. PMID 12662933. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2910754.
- ^ a b c d e f g Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN (June 2006). "Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis". J. Mol. Biol. 359 (4): 913–29. doi:10.1016/j.jmb.2006.04.004. PMC 2910082. PMID 16698036. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2910082.
- ^ a b Ozand PT, Nyhan WL, Barshop BA (2005). "Part Thirteen Lipid Storage Disorders: Tay-Sachs disease/hexosaminidase A deficiency". Atlas of metabolic diseases. London: Hodder Arnold. pp. 539–546. ISBN 0-340-80970-1.
- ^ Boles DJ, Proia RL (March 1995). "The molecular basis of HEXA mRNA deficiency caused by the most common Tay-Sachs disease mutation". Am. J. Hum. Genet. 56 (3): 716–24. PMC 1801160. PMID 7887427. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1801160.
- ^ Toleman CA, Paterson AJ, Kudlow JE (February 2006). "The histone acetyltransferase NCOAT contains a zinc finger-like motif involved in substrate recognition". J. Biol. Chem. 281 (7): 3918–25. doi:10.1074/jbc.M510485200. PMID 16356930.
- ^ Besley GT, Broadhead DM (April 1976). "Studies on human N-acetyl-Beta-d-hexosaminidase C separated from neonatal brain". Biochem. J. 155 (1): 205–8. PMC 1172820. PMID 945735. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1172820.
- ^ Gao Y, Wells L, Comer FI, Parker GJ, Hart GW (March 2001). "Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain". J. Biol. Chem. 276 (13): 9838–45. doi:10.1074/jbc.M010420200. PMID 11148210.
- ^ Forsythe ME, Love DC, Lazarus BD, Kim EJ, Prinz WA, Ashwell G, Krause MW, Hanover JA (August 2006). "Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer". Proc. Natl. Acad. Sci. U.S.A. 103 (32): 11952–7. doi:10.1073/pnas.0601931103. PMC 1567679. PMID 16882729. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1567679.
- ^ Gutternigg M, Rendić D, Voglauer R, Iskratsch T, Wilson IB (April 2009). "Mammalian cells contain a second nucleocytoplasmic hexosaminidase". Biochem. J. 419 (1): 83–90. doi:10.1042/BJ20081630. PMC 2850170. PMID 19040401. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2850170.
External links
- GeneReviews/NCBI/NIH/UW entry on hexosaminidase A deficiency, Tay-Sachs disease
- MeSH A hexosaminidase A
- EC 3.2.1.52
Hydrolase: sugar hydrolases (EC 3.2) 3.2.1: Glycoside hydrolases Cellulase · Alpha-glucosidase (Acid, Neutral AB, Neutral C) · Beta-glucosidase (cytosolic) · Debranching enzymeOtherAmylase (Alpha-Amylase) · Chitinase · Lysozyme · Neuraminidase (NEU1, NEU2, NEU3, NEU4, Bacterial neuraminidase, Viral neuraminidase) · Galactosidases (Alpha, Beta) · alpha-Mannosidase · Glucuronidase · Hyaluronidase · Pullulanase · Glucosylceramidase (lysosomal, non-lysosomal) · Galactosylceramidase · Alpha-N-acetylgalactosaminidase (NAGA) · Alpha-N-acetylglucosaminidase · Fucosidase · Hexosaminidase (HEXA, HEXB) · Iduronidase · Maltase-glucoamylase · Heparanase (HPSE2)3.2.2: Hydrolysing
N-Glycosyl compoundsAnabolism Catabolism Neuraminidase · Beta-galactosidase · Hexosaminidase · mannosidase (alpha-Mannosidase, beta-mannosidase) · Aspartylglucosaminidase · Fucosidase · NAGATransport M6P tagging Categories:- Genes on chromosome 15
- Genes on chromosome 5
- Genes on chromosome 10
- Genes on chromosome 17


Wikimedia Foundation. 2010.