- Alveolar macrophage
-
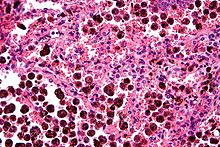 Micrograph showing hemosiderin-laden alveolar marcophages, as seen in a pulmonary haemorrhage. H&E stain.
Micrograph showing hemosiderin-laden alveolar marcophages, as seen in a pulmonary haemorrhage. H&E stain.
An alveolar macrophage (or dust cell) is a type of macrophage found in the pulmonary alveolus, near the pneumocytes, but separated from the wall.
Activity of the alveolar macrophage is relatively high, because they are located at one of the major boundaries between the body and the outside world.
Dust cells are another name for monocyte derivatives in the lungs that reside on respiratory surfaces and clean off particles such as dust or microorganisms.
Aveolar macrophages are frequently seen to contain granules of exogenous material such as particulate carbon that they have picked up from respiratory surfaces. Such black granules may be especially common in smoker's lungs or long-term city dwellers.
Inhaled air may contain particles or organisms which would be pathogenic. The respiratory pathway is a prime site for exposure to pathogens and toxic substances. The respiratory tree, comprising the larynx, trachea, and bronchioles, is lined by ciliated epithelia cells that are continually exposed to harmful matter [1]. When these insidious agents infiltrate our superficial barriers, the immune systems of our bodies respond in an orchestrated defense involving a litany of specialized cells which target the threat, neutralize it, and clean up the remnants of the battle.
Deep within the lungs exists its constituent alveoli sacs, the sites responsible for the uptake of oxygen and excretion of carbon dioxide. There are three major alveolar cell types in the alveolar wall (pneumocytes):
- Type I (Squamous Alveolar) cells that form the structure of an alveolar wall
- Type II (Great Alveolar) cells that secrete pulmonary surfactant to lower the surface tension of water and allows the membrane to separate, thereby increasing the capability to exchange gases. Surfactant is continuously released by exocytosis. It forms an underlying aqueous protein-containing hypophase and an overlying phospholipid film composed primarily of dipalmitoyl phosphatidylcholine.
- Macrophages that destroy foreign material, such as bacteria.
Type 1 and type 2 pneumocytes [2]. Type 1 pneumocytes (or membranous pneumocytes) form the structure of the alveolus and are responsible for the gas exchange in the alveolus [3]. Type 1 pneumocytes are squamous epithelial cells which are characterized by a superficial layer consisting of large, thin, scale-like cells; they also cover 95% of the alveolar surface, although they are only half as numerous as Type 2 pneumocytes[1][3]. Type 2 pneumocytes are important in that they can proliferate and differentiate into type 1 pneumocytes, which cannot replicate and are susceptible to a vast numbers of toxic insults [1]. Type 2 pneumocytes are also important because they secrete pulmonary surfactant(PS), which consists 80-90% of phospholipids [(phosophatidylcholine(PC), phosphatidyglycerol(PG), phosphaditylinositol (PI)] and 5-10% of surfactant proteins (SP-A, SP-B, SP-C, AND SP-D)[4]. PS is synthesized as lamellar bodies, which are structures consisting of closely packed bilayers that are secreted and then undergo transformation into a morphological form called tubular myelin [4]. PS plays an important role in maintaining normal respiratory mechanics by reducing alveolar surface tension. By lowering alveolar surface tension, PS reduces the energy required to inflate the lungs, and reduces the likelihood of alveolar collapse during expiration [4]. Loosely attached to these alveoli sacs are the alveolar macrophages that protect the lungs from a broad array of microbes and aerosols by devouring and ingesting them through phagocytosis [2].
Alveolar macrophages are phagocytes that play a critical role in homeostasis, host defense, the response to foreign substances, and tissue remodeling [2]. Since alveolar macrophages are pivotal regulators of local immunological homeostasis, their population density is decisive for the many processes of immunity in the lungs. They are highly adaptive components of the innate immune system and can be specifically modified to whatever functions needed depending on their state of differentiation and micro-environmental factors encountered. Alveolar macrophages release numerous secretory products and interact with other cells and molecules through the expression of several surface receptors. Alveolar macrophages are also involved in the phagocytosis of apoptic and necrotic cells that have undergone cell-death [3]. They must be selective of the material that is phagocytized because normal cells and structures of the body must not be compromised [3]. To combat infection, the phagocytes of the innate immune system facilitates many pattern recognition receptors (PRR) to help recognize pathogen-associated molecular patterns (PAMPs) on the surface of pathogenic microorganisms [5]. PAMPs all have the common features of being unique to a group of pathogens but invariant in their basic structure; and are essential for pathogenicity(ability of an organism to produce an infectious disease in another organism)[5]. Proteins involved in microbial pattern recognition include mannose receptor, complement receptors, DC-SIGN,Toll-like receptors(TLRs), the scavenger receptor, CD14, and Mac-1[5][6]. PRRs can be divided into three classes:
- signaling PRRs that activate gene transcriptional mechanisms that lead to cellular activation,
- endocytic PRRs that function in pathogen binding and phagocytosis, and
- secreted PRRs that usually function as opsonins or activators of complement.
The recognition and clearance of invading microorganisms occurs through both opsonin-dependent and opsonin–independent pathways. The molecular mechanisms facilitating opsonin-dependent phagocytosis are different for specific opsonin/receptor pairs. For example, phagocytosis of IgG-opsonized pathogens occurs through the Fcγ receptors (FcγR), and involves phagocyte extensions around the microbe, resulting in the production of pro-inflammatory mediators. Conversely, complement receptor-mediated pathogen ingestion occurs without observable membrane extensions (particles just sink into the cell) and does not generally results in an inflammatory mediator response.
Following internalization, the microbe is enclosed in a vesicular phagosome which then undergoes fusion with primary or secondary lysosomes, forming a phagolysosome[5]. There are various mechanisms that lead to intracellular killing; there are oxidative processes, and others independent of the oxidative metabolism. The former involves the activation of membrane enzyme systems that lead to a stimulation of oxygen uptake (known as the respiratory burst), and its reduction to reactive oxygen intermediates (ROIs), molecular species that are highly toxic for microorganisms [5]. The enzyme responsible for the elicitation of the respiratory burst is known as nicotinamide denine dinucleotide phosphate (NADPH) oxidase, which is composed of five subunits [5]. One component is a membrane cytochrome made up of two protein subunits, gp91phox and p22phox; the remaining three components are cytosolic-derived proteins: p40phox, p47phox, and p67phox [5]. NADPH oxidase exists in the cytosol of the AM when in a quiescent state; but upon activation, two of its cytosolic components, p47phox and p67phox, have their tyrosine and serine residues phosphorylated, which are then able to mediate translocation of NADPHox to the cytochrome component, gp91phox/p22phox, on the plasma membrane via cytoskeletal elements [5][7].
Compared to other phagocytes, the respiratory burst in AM is of a greater magnitude [5]. Oxygen-independent microbicidal mechanisms are based on the production of acid, on the secretion of lysozymes, on iron-binding proteins, and on the synthesis of toxic cationic polypeptides [5]. Macrophages possess a repertoire of antimicrobial molecules packaged within their granules and lysosomes [5]. These organelles contain a plethora of degradative enzymes and antimicrobial peptides that are released into the phagolysosome, such as proteases, nucleases, phosphatases, esterases, lipases, and highly basic peptides [5]. Moreover, macrophages possess a number of nutrient deprivation mechanisms that are used to starve phagocytosed pathogens of essential micronutrients [5]. Certain microorganisms have evolved countermeasures which enable them to evade being destroyed by phagocytes. Although lysosomal-mediated degradation is an efficient means by which to neutralize an infection and prevent colonization, several pathogens parasitize macrophages, exploiting them as a host cell for growth, maintenance and replication [5]. Parasites like Toxoplasma gondii and mycobacteria are able to prevent fusion of phagosomes with lysosomes, thus escaping the harmful action of lysosomal hydrolases. Others avoid lysosomes by leaving the phagocytic vacuole, to reach the cytosolic matrix where their development is unhindered. In these instances, macrophages may be triggered to actively destroy phagocytosed microorganisms by producing a number of highly toxic molecules and inducing deprivational mechanism to starve it [5]. Finally, some microbes have enzymes to detoxify oxygen metabolites formed during the respiratory burst [5].
When insufficient to ward off the threat, alveolar macrophages can release proinflammatory cytokines and chemokines to call forth a highly developed network of defensive phagocytic cells responsible for the adaptive immune response.
The lungs are especially sensitive and prone to damage, thus to avoid collateral damage to type 1 and type II pneumocytes, alveolar macrophages are kept in a quiescent state, producing little inflammatory cytokines and displaying little phagocytic activity, as evidenced by downregulated expression of the phagocytic receptor Macrophage 1 antigen (Mac-1) [2][8]. AMs actively suppress the induction of two of the immunity systems of the body: the adaptive immunity and humoral immunity. The adaptive immunity is suppressed through AM’s effects on interstitial dendritic cells, B-cells and T-cells, as these cells are less selective of what they destroy, and often cause unnecessary damage to normal cells. To prevent uncontrolled inflammation in the lower respiratory tract, alveolar macrophages secrete nitric oxide, prostaglandins, interleukin-4 and -10(IL-4, IL-10), and transforming growth factor-β (TGF-β) [8][9][10][11].
Contents
Nitric Oxide
NO is a major source of immunomodulation in rodents, and is produced by enzyme nitric oxide synthetase type 2 (NOS2) in the alveolar macrophage [10]. NO inhibits tyrosine phosphorylation of the kinases involved in production of the interleukin-2 (IL-2) receptor, the expression of which is fundamental for T cell proliferation [9]. In humans, however, NOS2 activity has been difficult to verify [10].
There are two explanations for the lack of responsiveness in the promoter of human inducible nitric oxide synthetase (iNOS) to NO activation by lipopolysaccharides (LPS) + interferon-gamma (IFN-γ) [10]. The first is that there are various inactivating nucleotide variations in the human counterpart of the enhancer element that regulates LPS/IFN-γ induced expression of the mouse NOS2 gene. The second is because of the absence of a nuclear factor in human macrophages that is required for optimum expression of gene NOS2 (LPS-inducible nuclear factor-kappa B/Rel complex) [10]. It is assumed that the difficulty in verifying NOS2 is due to a much more tightly controlled expression in human AMs as compared to that in the rodent AMs [10]. NOS2 is part of an autoregulatory feedback loop, wherein an allergen or provoker stimulates inflammatory cytokine production, which in turn stimulates NO production, and NO down-regulates cytokine production [10]. In rats, NO inhibits the granulocyte-macrophage colony-stimulating factor (GM-CSF)-mediated maturation of dendritic cells, and in humans it inhibits the TNF-alpha-mediated maturation of human dendritic cells, through cyclic GMP-dependent mechanisms [10]. NO prolongs the ability of human dendritic cells to internalize antigens at sites of inflammation, therefore modulating the beginning steps leading to antigen-specific immune responses [10].
NO production has been implicated as relevant to the pathology of asthma. Patients with asthma show an increased expression of iNOS in airway epithelial cells and an increased level of nitric oxide in exhaled air [10].
Prostaglandin endoperoxide 2 (PGE2)
Many other immunomodulating factors have been isolated, the most important of which are prostaglandins and cytokines. PGE2 was the first immunomodulator to be derived from macrophages and described [10]. PGE2 functions in amplifiying peripheral blood lymphocyte IL-10 transcription and protein production; as well as in deactivating macrophages and T-cells [10]. PGE2 is an immunomodulatory eicosanoid derived from the cell membrane component, arachidonic acid, and is processed in the arachidonic acid cascade: the successive oxygenation and isomerization of arachidonic acid by cyclooxygenase and PGE2 synthase enzymes [12]. The regulation of target cells by PGE2 occurs via signaling through four cell membrane-associated G-protein-coupled E-prostanoid (EP) receptors, named EP1, EP2, EP3, and EP4 [12]. PGE2 inhibits bacterial killing and ROI production by AM by impairing Fcγ-mediated phagocytosis through its ability to stimulate the production of intracellular cyclic adenosine monophosphate (cAMP) effectors via EP2 and EP4 receptors signaling [7][12]. EP2 and EP4 receptors signal primarily through stimulatory G protein (Gs), increasing adenylyl cyclase (AC) activity and subsequent cAMP formation [7]. cAMP is a second messenger that influences multiple cellular functions via the activation of two downstream effector molecules, protein kinase A (PKA) and the exchange proteins directly activated by cAMP (Epac-1 and -2)[7]. Epac-1 and PKA are both important factors involved in the inhibition of AM bacterial killing [7]. The effects of PKA results from its ability to phosophorylate serine and threonine residues on many cellular proteins, especially transcription factor cAMP response element binding protein (CREB). cAMP/PKA/CREB axis mediates the inhibition of TNF-alpha release [7]. The killing of phagocytosed bacteria by AMs is dependent upon several distinct microbicidal mechanisms, like the reduced NADPH oxidase-mediated release of ROI [5][7]. ROI generation by NADPH oxidase is an important bactericidal mechanism after FcR-mediated phagocytosis [7]. PGE2 activates both Gs-coupled EP2 and EP4 receptors by ligation, stimulating cAMP production and subsequent activation of downstream cAMP effectors, PKA and Epac-1; both which in turn impair the phosphorylation and phagosomal membrane translocation of NADPH oxidase component, p47phox, thereby inhibiting the respiratory burst [7].
Interleukin-4 and -10
IL-4 is a pleiotropic cytokine that plays a key role in the development of T helper type 2(Th2) cells. IL-4 is important for the differentiation of naïve CD4-T cells into mature Th2 type cells; as well as for Immunoglobulin (Ig) class switching to IgE and IgG4 during the development of immune responses [13][14]. Ig is a class of antibody found only in mammals that plays an important role in allergy response and defense against many kinds of pathogens by protecting the body against them by complement activation, opsonization for phagocytosis, and neutralization of their toxins [14].
IL-4 and IL-10 have both been shown to reduce the production of metalloproteinases (endopeptidases which break down collagen and other extracellular proteins) by human AMs [10][11]. IL-4 has dual effects upon macrophage biological function, which may be either stimulatory or inhibitory [11]. It enhances MHC class II antigen (extracellular protein complex that interacts exclusively with CD4-T cells as part of the exogenous pathway) and Mac-1(surface receptor as part of innate complement system) expression, thus promoting phagocytosis [11]. IL-4 has also been shown to inhibit the production of PGE2 by reducing the expression of the enzyme, prostaglandin H synthase -2 (PGHS-2), which is critical in the production of PGE2 [10]. However, IL-4 inhibits production of TNF-alpha, IL-1 and -6, which are all important cytokines in the proinflammatory response) [11].
IL-10 inhibits the secretion of pro-inflammatory cytokines TNF-alpha and INF-gamma, thus suppressing the proliferation of T-cells, NK cells, and AM [10]. IL-10 shares similar immunomodulating mechanisms to TGF-β [10]. It is thought that both cytokines reduce the rate of apoptosis in human alveolar macrophages, thus indirectly enhancing alveolar macrophage-mediated inhibition of T-cell proliferation [10]. There is a significant increase in the basal rate of apoptosis upon activation by bacterial products. Apoptosis is particularly regulated by the presence of cytokines: IFN-γ increases the rate of apoptosis, whereas IL-10 and TGF-β decrease it [10]. However, IL-10 has counterproductive effects on the immune system, and has been shown to actually promote infection by foreign pathogens. The role of IL-10 in bacterial and parasitic infection has been discovered as a strategy to evade host immune systems [4]. There are bacteria which parasitize AMs by invading through their membranes, and thrive by growing and replicating inside of them, exploiting AMs as host cells. Normally, this infection can be eliminated by T-cells, which activate enzymes in alveolar macrophages that destroy the bacteria; but these bacteria have been shown to alter the cytokine signaling network to their advantage. As an inhibitory cytokine, IL-10 facilitates the infection of human alveolar macrophages and monocytes by completely reversing the protective effect of IFN-γ against intracellular Legionella pneumophila replication [4]. Yersinia enterocolitica has also been shown to releases virulence antigen LcrV, which induces IL-10 through Toll-like receptor-2 and CD14 (an accessory surface protein of TLR4-mediated LPS-signaling), resulting in the suppression of IFN-γ and TNF-alpha suppression [4].
Transforming growth factor β (TGF-β)
In normal conditions, alveolar macrophages adhere closely to alveolar epithelial cells, thus inducing the expression of the αvβ6 integrin. Integrins are dimeric cell-surface receptors composed of alpha and beta subunits, which activates TGF-β [15][16]. TGF-β is a multifunctional cytokine that modulates a variety of biological processes such as cell growth, apoptosis, extracellular matrix synthesis, inflammation, and immune responses [17]. TGF-β tightly regulates anti-inflammatory activity by suppressing pro-inflammatory cytokine production, thereby inhibiting T-lymphocyte function [18]. Integrins avβ6 and avβ8 sequester latent TGF-β to the cell surface, where activation can be tightly coupled to cellular responses to environmental stress in the maintenance of homeostasis; integrins also localize activated TGFβ in the vicinity of the macrophages [19]. Normally mature TGFβ is secreted as a latent complex with its N-terminal fragment, latency-associated peptide (LAP), which inhibits its activity [17]. The latent complex is covalently linked to the extracellular matrix by binding to latent TGF-β-binding proteins [15]. TGF-β is activated by diverse mechanisms in the lung, ultimately involving either proteolysis or conformational alteration of the LAP[19]. αvβ6 integrin is able to mediate activation of TGF-β by binding to TGF-β1 LAP, which serves as a ligand binding site for the integrin, and is an essential component of the TGF-β activation apparatus[17][20]. Once activated, TGFβ leads to the suppression of macrophage functionality (cytokine production and phagocytosis)[17]. Binding of activated TGF-β to its receptors expressed on alveolar macrophages induces a downstream signaling cascade, including phosphorylation of receptor-regulated Small Mothers Against Decapentaplegic (R-SMAD)homologs 2 and 3 [2][17][18]. Phosphorylated SMAD-2 and -3 then form heteromeric complexes with common-mediator SMAD 4 (co-SMAD-4). Once assembled, the complexes translocates into the nucleus via the nuclear pore with the assistance of importins alpha/beta. Once in the nucleus, these complexes accumulate and eventually act as a transcription factors, regulating the expression of TGF-β target genes [18]. Thus TGF-β signaling involves a direct pathway from the receptors on the surface of a cell to the nucleus.
Releasing the Brakes on Alveolar Macrophages
Toll-like receptors (TLRs) are signaling PRRs, a family of receptors that is capable of recognizing conserved microbial patterns like components of the bacterial cell wall, microbial nucleic acids, and bacterial motility [6]. Although bacteria have evolved means of evading host defense mechanisms, they express PAMPs, such as lipoglycans and lipoproteins that are recognized by cells of the innate immune system through the TLRs [6]. Upon detection of a dangerous antigen by interaction between TLR and PAMPs, TLR triggers inflammatory and defensive responses in the host by inducing actin polymerization in alveolar macrophages (a crucial component in endocytosis and motility) [17]. Actin polymerization in alveolar macrophages causes the suppression of integrin expression, which in turn causes the deactivation of TGF-β and the downregulation of the basal phosphorylation level of SMAD 2/3; subsequently leading to the activation and detachment from the alveolar epithelial cells of alveolar macrophages [17][15]. Upon releasing the brakes of macrophage activation, macrophages become primed for phagocytosis and begin to secrete proinflammatory cytokines (TNF-α and IL-6) [17].
The priming of macrophages involves the enhancement of respiratory burst activity by IFN- γ and TNF-α [5]. IFN- γ induces both an increased affinity of the NADPH oxidase for NADPH in macrophages, as well as an increased rate of gene transcription and message expression for gp91phox protein [5]. TNF-α acts as an autocrine stimulus by increasing the expression of both p47phox and p67phox transcripts. The ROIs produced during the respiration burst response, in turn, enhance production of TNF-α by macrophages[5].
Halting the Activated Alveolar Macrophage Gas exchange must be restored as quickly as possible to avoid collateral damage, so activated lymphocytes secrete IFN- γ to stimulate the production of matrix metalloproteinase MMP-9 by macrophages [17]. AMs have been reported to produce MMP-9 partly via PGE2-dependent PKA signaling pathways, which are the pathways involved in the inhibition of phagocytosis [21]. MMP-9 activates latent TGF-β, reinducing expression of αvβ6 on alveolar epithelial cells, thereby putting the brakes back on AMs [2][17][21]. Activation of TGF-β is also advantageous because its production stimulates collagen synthesis in interstitial fibroblasts, which is necessary for restoring alveolar wall architecture [17].
Recent Research into Alveolar Macrophages and Future Prospects Research into AM functionality has been on the rise since AMs are one of the first lines of a defense against invasive pathogens. One of the most prominent fields is investigating liposomes as deliverers of antibiotics for treatment of respiratory intracellular infections. Intracellular parasites, such as M. tuberculosis, C. pneumoniae, L. monocytogenes, L. pneumophila, and F. tularensis, (to name a few) are taken up by AMs via phagocytosis, but are resistant to the biocidal mechanisms of AMs and can survive intracellularly, thus inducing severe respiratory infections [22]. Pulmonary tuberculosis is caused by M. tuberculosis, and is now a major infectious disease worldwide and its incidence is increasing, especially in association with the AIDS pandemic [22]. For sterilization of intracellular parasites in AMs, antibiotics are normally given orally or intravenously, but much of the antibiotics disperse to many different tissues, diminishing its effectiveness. Pulmonary administration of mannosylated lyposomes is a much more direct, efficient route in targeting AMs; it enhances antimicrobial effect, reduces the dosage needed, and avoids unnecessary distribution to the blood [22][23]. Since mannose receptors are exclusively expressed on the surface of AM, mannosylation of liposomes is an appealing approach to cell-selective targeting to AM [23]. The efficacy of pulmonary administration of ciprofloxacin (CPFX) incorporated into mannosylated liposomes (mannosylated CPFX-lipososomes) was examined in rats, and determined to be an efficient means to target AMs [22].
References
- ^ a b c Hussain, Aliya N. “Immune system of the lungs”. Pathologic basis of disease7 (2006): Chapter 15
- ^ a b c d e f Lambrecht, B. N. "Alveolar Macrophage in the Driver's Seat." Immunity 24.4 (2006): 366-8.
- ^ a b c d Guyton, Arthur C. “Physiology of the respiratory system.” Textbook of Medical Physiology 11(2007): Chapter 33, 431-433
- ^ a b c d e f oshizawa, S., et al. "Legionella Pneumophila Evades Gamma Interferon-Mediated Growth Suppression through Interleukin-10 Induction in Bone Marrow-Derived Macrophages." Infection and immunity 73.5 (2005): 2709-17.
- ^ a b c d e f g h i j k l m n o p q r s t Stafford, J. L., N. F. Neumann, and M. Belosevic. "Macrophage-Mediated Innate Host Defense Against Protozoan Parasites." Critical reviews in microbiology 28.3 (2002): 187-248.
- ^ a b c Krutzik, Stephan R., and Robert L. Modlin. "The Role of Toll-Like Receptors in Combating Mycobacteria." Seminars in Immunology, 16.1 (2004): 35-41.
- ^ a b c d e f g h i Serezani, C. H., et al. "Prostaglandin E2 Suppresses Bacterial Killing in Alveolar Macrophages by Inhibiting NADPH Oxidase." American journal of respiratory cell and molecular biology 37.5 (2007): 562-70.
- ^ a b Holt, P. G., et al. "Downregulation of the Antigen Presenting Cell Function(s) of Pulmonary Dendritic Cells in Vivo by Resident Alveolar Macrophages." The Journal of experimental medicine 177.2 (1993): 397-407.
- ^ a b BUNN, H. J., C. R. A. HEWITT, and J. GRIGG. "Suppression of Autologous Peripheral Blood Mononuclear Cell Proliferation by Alveolar Macrophages from Young Infants." Clinical & Experimental Immunology 128.2 (2002): 313-7.
- ^ a b c d e f g h i j k l m n o p q r Bingisser, R. M., and P. G. Holt. " Swiss medical weekly : official journal of the Swiss Society of Infectious Diseases, the Swiss Society of Internal Medicine, the Swiss Society of Pneumology 131.13-14 (2001): 171-9.
- ^ a b c d e Lacraz, S., et al. "Suppression of Metalloproteinase Biosynthesis in Human Alveolar Macrophages by Interleukin-4." The Journal of clinical investigation 90.2 (1992): 382-8.
- ^ a b c Brock, Thomas G., et al. "Effects of Prostaglandin E2 on the Subcellular Localization of Epac-1 and Rap1 Proteins during Fcγ-Receptor-Mediated Phagocytosis in Alveolar Macrophages." Experimental Cell Research, 314.2 (2008): 255-63.
- ^ Pouliot, P., et al. "Interleukin-4 Production by Human Alveolar Macrophages." Clinical & Experimental Allergy 35.6 (2005): 804-10.
- ^ a b Paul, W. E. "Interleukin-4: A Prototypic Immunoregulatory Lymphokine." Blood 77.9 (1991): 1859-70.
- ^ a b Araya, J., et al. "Integrin-Mediated Transforming Growth Factor-Beta Activation Regulates Homeostasis of the Pulmonary Epithelial-Mesenchymal Trophic Unit." The American journal of pathology 169.2 (2006): 405-15
- ^ David G. Morris, et al. "Loss of Integrin v6-Mediated TGF- Activation Causes Mmp12-Dependent Emphysema." Nature 422 (2003): 169-73.
- ^ a b c d e f g h i j k Takabayshi, Kenji, et al. "Induction of a Homeostatic Circuit in Lung Tissue by Microbial Compounds." Immunity, 24.4 (2006): 475-87.
- ^ a b c RAY, CHAD A., et al. "Transforming Growth Factor-Beta Activation and Signaling in the Alveolar Environment during Pneumocystis Pneumonia." The Journal of eukaryotic microbiology 53 (2006): S127-9.
- ^ a b Annes, J.P., et al. “Making sense of latent TGFbeta activation.” Journal of Cell Science, 116 (2003): 217-224
- ^ Munger, John S., et al. "A Mechanism for Regulating Pulmonary Inflammation and Fibrosis: The Integrin αvβ6 Binds and Activates Latent TGF β1." Cell, 96.3 (1999): 319-28.
- ^ a b Ohbayashi, H., and K. Shimokata. "Matrix Metalloproteinase-9 and Airway Remodeling in Asthma." Current drug targets.Inflammation and allergy 4.2 (2005): 177-81.
- ^ a b c d Chono, Sumio, et al. "Efficient Drug Targeting to Rat Alveolar Macrophages by Pulmonary Administration of Ciprofloxacin Incorporated into Mannosylated Liposomes for Treatment of Respiratory Intracellular Parasitic Infections." Journal of Controlled Release, 127.1 (2008): 50-8.
- ^ a b Wijagkanalan, Wassana, et al. "Efficient Targeting to Alveolar Macrophages by Intratracheal Administration of Mannosylated Liposomes in Rats." Journal of Controlled Release, 125.2 (2008): 121-30.
External links
- alveolar+macrophage at eMedicine Dictionary
- Histology at BU 13906loa - "Respiratory System: lung (human), alveolar macrophages"
- Histology at KUMC resp-resp16 "Alveoli"
- Slide at ufl.edu
Myeloid lineage - Blood (WBC and RBC) Cellular/
HSCsCFU-GMHistiocytes · Kupffer cells · Alveolar macrophage · Microglia · Osteoclasts · Epithelioid cells · giant cells (Langhans giant cells, Foreign-body giant cell, Touton giant cells)CFU-DLCommonCFU-BasoCFU-EosCFU-MegCFU-ECFU-MastNoncellular Human cell types / list derived primarily from mesoderm Paraxial muscle: Myoblast → Myocyte · Myosatellite cell · Tendon cell · Cardiac muscle cell
adipose: Lipoblast → AdipocyteDigestive systemIntermediate Urinary system (RSC)Angioblast → Endothelial cell · Mesangial cell (Intraglomerular, Extraglomerular) · Juxtaglomerular cell · Macula densa cell
Stromal cell → Interstitial cell → Telocytes
Simple epithelial cell → Podocyte · Kidney proximal tubule brush border cellLateral plate/
hemangioblastsee lymphocytessee myeloid cellsCategories:- Macrophages
- Phagocytes
{kind=link}
Wikimedia Foundation. 2010.