- Monte Carlo methods for electron transport
-
The Monte Carlo method for electron transport is a semiclassical Monte Carlo(MC) approach of modeling semiconductor transport. Assuming the carrier motion consists of free flights interrupted by scattering mechanisms, a computer is utilized to simulate the trajectories of particles as they move across the device under the influence of an electric field using classical mechanics. The scattering events and the duration of particle flight is determined through the use of random numbers.
Contents
Background
Boltzmann transport equation
The Boltzmann transport equation model has been the main tool used in the analysis of transport in semiconductors. The BTE equation is given by:
The distribution function, f, is a dimensionless function which is used to extract all observable of interest and gives a full depiction of electron distribution in both real and k-space. Further, it physically represents the probability of particle occupation of energy k at position r and time t. In addition, due to being a seven-dimensional integro-differential equation (six dimensions in the phase space and one in time) the solution to the BTE is cumbersome and can be solved in closed analytical form under very special restrictions. Numerically, solution to the BTE is employed using either a deterministic method or a stochastic method. Deterministic method solution is based on a grid-based numerical method such as the spherical harmonics approach, whereas the Monte Carlo is the stochastic approach used to solve the BTE.
Monte Carlo method
The semiclassical Monte Carlo method is a statistical method used to yield exact solution to the Boltzmann transport equation which includes complex band structure and scattering processes. This approach is semiclassical for the reason that scattering mechanisms are treated quantum mechanically using the Fermi's Golden Rule, whereas the transport between scattering events is treated using the classical particle notion. The Monte Carlo model in essence tracks the particle trajectory at each free flight and chooses a corresponding scattering mechanism stochastically. Two of the great advantages of semiclassical Monte Carlo are its capability to provide accurate quantum mechanical treatment of various distinct scattering mechanisms within the scattering terms, and the absence of assumption about the form of carrier distribution in energy or k-space. The semiclassical equation describing the motion of an electron is
where F is the electric field, E(k) is the energy dispersion relation, and k is the momentum wave vector. To solve the above equation, one needs strong knowledge of the band structure (E(k)). The E(k) relation describes how the particle moves inside the device, in addition to depicting useful information necessary for transport such as the density of states (DOS) and the particle velocity. A Full-band E(K) relation can be obtained using the semi-empirical pseudopotential method.[1]
Hydrodynamic and drift diffusion method
Both drift diffusion (DD) and the hydrodynamic (HD) models can be derived from the moments of the Boltzmann transport equation (BTE) using simplified approximation valid for long channel devices. The DD scheme is a the most classical approach and usually solves the Poisson equation and the continuity equations for carriers considering the drift and diffusion components. In this approach, the charge transit time is assumed to be very large in comparison to the energy relaxation time.[2] On the other hand, the HD method solves the DD scheme with the energy balance equations obtained from the moments of the Boltzmann Transport Equation (BTE).[3][4] Thus, one may capture and calculate physical details such as carrier heating and the velocity overshoot effect. Needless to say, an accurate discretization method is required in HD simulation, since the governing equations are strongly coupled and one has to deal with larger number of variables compared to the DD scheme.
Comparison of semiclassical models
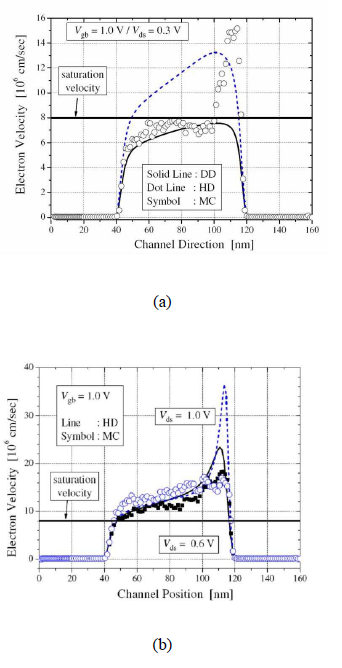
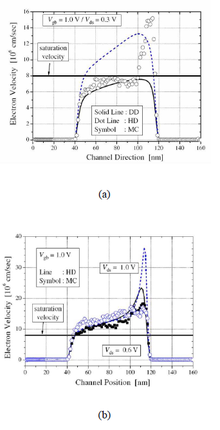 Average carrier velocity for a 80nm nmos comparing the various semiclassical simulation model (a) Vds= 0.3V (b) Vds= 0.6V
Average carrier velocity for a 80nm nmos comparing the various semiclassical simulation model (a) Vds= 0.3V (b) Vds= 0.6V
The accuracy of semiclassical models are compared based on the BTE by investigating how they treat the classical velocity overshoot problem, a key short channel effect (SCE) in transistor structures. Essentially, velocity overshoot is a nonlocal effects of scaled devices, which is related to the experimentally observed increase in current drive and transconductance.[5] As the channel length becomes smaller, the velocity is no longer saturated in the high field region, but it overshoots the predicted saturation velocity. The cause of this phenomenon is that the carrier transit time becomes comparable to the energy relaxation time, and therefore the mobile carriers do not have enough time to reach equilibrium with the applied electric field by scattering in the short channel devices.[6] The summary of simulation results (Illinois Tool: MOCA) with DD and HD model is shown in figure beside. In the figure (a), the case when the field is not high enough to cause the velocity overshoot effect in the whole channel region is shown. Note that at such limit, the data from the DD model fit well to the MC model in the non-overshoot region, but the HD model overestimate the velocity in that region. The velocity overshoot is observed only near the drain junction in the MC data and the HD model fits well in that region. From the MC data, it can be noticed that the velocity overshoot effect is abrupt in the high-field region, which is not properly included in the HD model. For high field conditions as shown in the figure (b) the velocity overshoot effect almost all over the channel and the HD results and the MC results are very close in the channel region.
Monte Carlo for semiconductor transport
Band structure
Band structure describes the relationship between energy(E) and wave vector(k). The band structure is used to compute the movement of carriers under the action of the electric field, scattering rate, and final state after the collision. Silicon band structure and its Brillouin zone are shown in figure below, but there is no analytical expression which satisfies entire Brillouin zone. By using some approximation, there are two analytical models for band structure, namely the parabolic and the non-parabolic modes.

Parabolic band structure
For the concept of band structure, parabolic energy bands are generally assumed for simplicity. Electrons reside, at least when close to equilibrium, close to the minima of the E(k) relation. Then the E(k) relation can be extended in a Taylor series as
Because the first derivative vanishes at the band minimum, so the gradient of E(k) is zero at k = 0. Thus,
which yields the definition of the effective mass tensor
This expression is true for semiconductor which has isotropic effective mass, for instance GaAs. In case of silicon, conduction band minima does not lie at k = 0 and the effect mass depends on the crystallographic orientation of the minimum as
where
 describe longitudinal and transverse effective mass, respectively.
describe longitudinal and transverse effective mass, respectively.Non-parabolic band structure
For higher applied fields, carriers reside above the minimum and the dispersion relation, E(k), does not satisfy the simple parabolic expression described above. This non-parabolicity is generally described by
where α is a coefficient of non-parabolicity given by
where m0 is the electron mass in vacuum, and Eg is the energy gap.[7]
Full band structure
For many applications, non-parabolic band structure provides reasonable approximation. However, in case of very high field transport, which requires the better physical model of the full band structure. For full band approach, numerically generated table of E(k) is used. Full band approach for Monte Carlo simulation was first used by Karl Hess at the University of Illinois at Urbana-Champaign. This approach is based on empirical pseudopotential method suggested by Cohen and Bergstresser [18]. Full band approach is computationally expensive, however, following the advancement of the computational power, it can be used as a more general approach.[8]
Types of Monte Carlo simulation
One-particle Monte Carlo
For this type of simulation, one carrier is injected and the motion is tracked in the domain, until it exits through contact. Another carrier is then injected and the process repeated to simulate an ensemble of trajectories. This approach is mostly useful to study bulk properties, like the steady state drift velocity as a function of field.
Ensemble Monte Carlo
Instead of single carrier, a large ensemble of carriers is simulated at the same time. This procedure is obviously a good candidate for super-computation, since one may apply parallelization and vectorization. Also, it is now possible to perform ensemble averages directly. This approach is suitable for transient simulations.
Self-consistent ensemble Monte Carlo
This method couples the ensemble Monte Carlo procedure to Poisson’s equation, and is the most suitable for device simulation. Typically, Poisson’s equation is solved at fixed intervals to update the internal field, to reflect the internal redistribution of charge, due to the movement of carriers.
Random flight selection
The probability that the electron will suffer its next collision during dt around t is given by
where P[k(t)]dt is the probability that an electron in the state k suffers a collision during the time dt. Because of the complexity of the integral at the exponent, it is impractical to generate stochastic free flights with the distribution of the equation above. In order to overcome this difficulty, people use fictitious “self-scattering” scheme. By doing this, total scattering rate including this self-scattering, is constant and equal to, say, Γ. By random selection, if self-scattering is selected, k′ after the collision is same with k and the carrier continues flight without perturbation. Introducing a constant
 , the above equation reduces to
, the above equation reduces toRandom numbers r can be used very simply to generate stochastic free flights, which duration will then be given by tr = − τ0ln(r). The computer time used for self-scattering is more than compensated for by the simplification of the calculation of the free-flight duration.[9] To enhance the speed of free flight time calculation, several schemes such as “Constant Technique”, and “Piecewise Technique” are used to minimize the self-scattering events.
Scattering mechanisms
General background in solid-state physics
Important charge transport properties of semiconductor devices such as the deviance from Ohm’s law and the saturation of carriers mobility are a direct consequence of scattering mechanisms. It is thus of great importance for a semiconductor device simulation to capture the physics of such mechanisms. The semiconductor Monte Carlo simulation, in this scope, is a very powerful tool for the ease and the precision with which an almost exhaustive array of cattering mechanisms can be included. The duration of the free flights is determined from the scattering rates. At the end of each flight, the appropriate scattering mechanism must be chosen in order to determine the final energy of the scattered carrier, or equivalently, its new momentum and scattering angle. In this sense, one will distinguish two broad types of scattering mechanisms which naturally derive form the classic kinetic theory of collision between two bodies:
- • Elastic scattering, where the energy of the particle is conserved after being scattered. Elastic scattering will hence only change the direction of the particle’s momentum. Impurity scattering and surface scattering are, with a fair approximation, two good examples of elastic scattering processes.
- • Inelastic scattering, where energy is transferred between the scattered particle and the scattering center. Electronphonon interactions are essentially inelastic since a phonon of definite energy is either emitted or absorbed by the scattered particle.
Before characterizing scattering mechanisms in greater mathematical details, it is important to note that when running semiconductor Monte Carlo simulations, one has to deal mainly with the following types of scattering events [9]:
- • Acoustic Phonon: The charge carrier exchanges energy with an acoustic mode of the vibration of atoms in the crystal lattice. Acoustic Phonons mainly arise from thermal excitation of the crystal lattice.
- • Polar Optical: The charge carrier exchanges energy with one of the polar optical modes of the crystal lattice. These modes are not present in covalent semiconductors. Optical phonons arise from the vibration against each other of atoms of different types when there is more than one atom in the smallest unit cell, and are usually excited by light.
- • Non-Polar Optical: Energy is exchanged with an optical mode. Non-polar optical phonons must generally be considered in covalent semiconductors and the L-valley of GaAs.
- • Equivalent Intervalley Phonon: Due to the interaction with a phonon, the charge carrier transitions from initial states to final states which belong to different but equivalent valleys. Typically, this type of scattering mechanism describes the transition of an electron from one X-valley to another X-valley, or from one L-valley to another L-valley.[10]
- • Non Equivalent Intervalley Phonon: Involves the transition of a charge carrier between valleys of different types.
- • Piezoelectric Phonon: For low temperatures.
- • Ionized Impurity: Reflects the deviation of a particle from it ballistic trajectory due to Coulomb interaction with an ionized impurity in the crystal lattice. Because the mass of an electron is relatively small in comparison to the one of an impurity, the Coulomb cross section decreases rapidly with the difference of the modulus of momentum between the initial and final state.[9] Therefore impurity scattering events are mostly considered for intravalley scattering, intraband scattering and, to a minor extent, interband scattering.
- • Carrier-Carrier: (electron-electron, hole-hole and electron-hole interactions). When carrier concentration is high, this type of scattering reflects the electrostatic interaction between charge carriers. This problem becomes very quickly computationally intensive with an increasing number of particles in an ensemble simulation. In this scope, Particle-Particle–Particle-Mesh (P3M) algorithms, which distinguish short range and long range interaction of a particle with its surrounding charge gas, have proved efficient in including carrier-carrier interaction in the semiconductor Monte Carlo simulation.[11] Very often, the charge of the carriers is assigned to a grid using a Cloud-in-Cell method, where part of the charge of a given particle is assigned to a given number of closest grid points with a certain weight factor.
- • Plasmon: Reflects the effect of the collective oscillation of the charge carriers on a given particle.
Inclusion of scattering mechanisms in Monte Carlo
A computationally efficient approach to including scattering in Monte Carlo simulation consists in storing the scattering rates of the individual mechanisms in tables. Given the different scattering rates for a precise particle state, one may then randomly select the scattering process at the end of the free flight.
These scattering rates are very often derived using the Born approximation, in which a scattering event is merely a transition between two momentum states of the carrier involved. As discussed in section II-I, the quantum manybody problem arising from the interaction of a carrier with its surrounding environment (phonons, electrons, holes, plasmons, impurities,...) can be reduced to a two-body problem using the quasiparticle approximation, which separates the carrier of interest from the rest of the crystal.[9] Within these approximations, Fermi's Golden Rule gives, to the first order, the transition probability per unit time for a scattering mechanism from a state to a state
to a state  :
:where H' is the perturbation hamiltonian representing the collision and E and E′ are respectively the initial and final energies of the system constituted of both the carrier and the electron and phonon gas. The Dirac δ-function stands for the conservation of energy. In addition, the term
 , generally referred to as the matrix element, mathematically represents an inner product of the initial and final wave functions of the carrier [12]:
, generally referred to as the matrix element, mathematically represents an inner product of the initial and final wave functions of the carrier [12]:In a crystal lattice, the wavefunctions ψk(r) and ψk'(r) are simply Bloch waves. When it is possible, analytic expression of the Matrix elements are commonly found by Fourier expanding the hamiltonian H', as in the case of Impurity scattering [13] or acoustic phonon scattering.[14]
In the important case of a transition from an energy state E to an energy state E' due to a phonon of wave vector q and frequency ωq, the energy and momentum change is:where R is a reciprocal lattice vector. Umklapp processes (or U-processes) change the momentum of the particle after scattering and are therefore limiting the conduction in semiconductor crystals. Physically, U-processes occur when the final momentum of the particle points out of the first Brillouin zone. Once one knows the scattering probability per unit time from a state k to a state k', it is interesting to determine the scattering rate for a given scattering process. The scattering rate gives the probability per unit time to scatter from a state k to any other state in the reciprocal space. Therefore the scattering rate is
λ(k) = ∑ S(k,k') k' which can be readily used to determine the free flight time and the scattering process as discussed in section 3-3. It is important to note that this scattering rate will be dependent on the band structure of the material (the dependence arises from the matrix elements).
Selection of scattering mode and scattered trajectory
At the end of a free flight, a scattering mode and angle must be randomly chosen. In order to determine the scattering mechanism, one has to consider all the scattering rates λ1,λ2,...,λn of the mechanisms relevant to the simulation as well as the total scattering rate at the time of scattering
λtot(tsc) = ∑ λi. i Selecting a scattering mechanism then simply results in generating a uniformly distributed random number 0 < r < 1 and referring to the following rules
A computationally efficient approach to selecting the scattering mechanism consists in adding a “void” scattering mechanism so that λtot remains constant over time. If a particle is scattered according to this mechanism, it will keep its ballistic trajectory after scattering takes place. In order to chose a new trajectory, one must first derive the energy (or momentum) of the particle after scattering
where the term
 accounts for phonon emission or absorption and the term ΔEC is non-null for inter-valley scattering. The final energy (and the band structure) directly yield the modulus of the new momentum k'. At this point on only needs to chose a new direction (or angle) for the scattered particle. In some simple cases as phonon scattering and a parbolic dispersion relation, the scattering angle is random and evenly distributed on the sphere of radius k'. Using spherical coordinates, the process of chosing the angle is equivalent to randomly picking two angles θand ψ. If the angle is distributed with a distribution p(θ,ψ), then for a uniform distribution of angles, the probability to pick a point of the sphere is
accounts for phonon emission or absorption and the term ΔEC is non-null for inter-valley scattering. The final energy (and the band structure) directly yield the modulus of the new momentum k'. At this point on only needs to chose a new direction (or angle) for the scattered particle. In some simple cases as phonon scattering and a parbolic dispersion relation, the scattering angle is random and evenly distributed on the sphere of radius k'. Using spherical coordinates, the process of chosing the angle is equivalent to randomly picking two angles θand ψ. If the angle is distributed with a distribution p(θ,ψ), then for a uniform distribution of angles, the probability to pick a point of the sphere isIt is possible, in this case, to separate the two vaariables. Integrating over ψ then over θ, one finds
The two spherical angles can then be chosen, in the uniform case, by generating two random numbers 0 < r1, r2 < 1 such that
Quantum corrections for Monte Carlo simulation
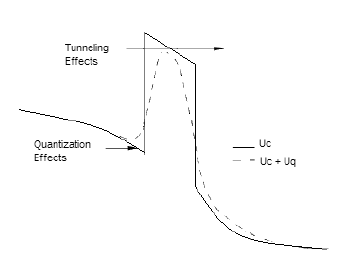
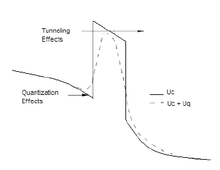 Effects Quantum Correction
Effects Quantum CorrectionThe current trend of scaling down semiconductor devices has forced physicists to incorporate quantum mechanical issues in order to acquire a thorough understanding of device behavior. Simulating the behavior of nano-scale devices necessitates the use of a full quantum transport model especially for cases when the quantum effects cannot be ignored. This complication, however, can be avoided in the case of practical devices like the modern day MOSFET, by employing quantum corrections within a semi-classical framework. The semi-classical Monte Carlo model can then be employed to simulate the device characteristics. The quantum corrections can be incorporated into a Monte Carlo simulator by simply introducing a quantum potential term which is superimposed onto the classical electrostatic potential seen by the simulated particles. Figure beside pictorially depicts the essential features of this technique. The various quantum approaches available for implementation are described in the following subsections.
Wigner-based correction
The Wigner transport equation forms the bases for the Wigner-based quantum correction.
where, k is the crystal momentum, V is the classical potential, the term on the RHS is the effect of collision,the fourth term on the LHS represents non-local quantum mechanical effects. The standard Boltzmann Transport Equation is obtained when the non-local terms on the LHS disappear in the limit of slow spatial variations. The simplified (for α = 0) quantum corrected BTE then becomes
where the quantum potential is contained in the term Vω.
Effective potential correction
This method of quantum correction was developed by Feynman and Hibbs in 1965. In this method the effective potential is derived by calculating the contribution to the path integral of a particle’s quantum fluctuations around its classical path. This calculation is undertaken by a variational method using a trial potential to first order. The effective classical potential in the average point on each path then becomes
Schrödinger-based correction
This approach involves periodical solving of a Schrödinger equation in a simulation with the input being the selfconsistent electrostatic potential. The exact energy levels and wavefunctions relating to the electrostatic potential solution are employed to calculate the quantum potential. The quantum correction obtained on the bases of this method can be visualised by the following equation
where Vschr is the quantum correction potential, z is the direction perpendicular to the interface, nq is the quantum density from the Schrödinger equation which is equivalent to the converged Monte Carlo concentration, Vp is the potential from the Poisson solution, V0 is the arbitrary reference potential far away from the quantum region such that the correction goes to null in the region of semi-classical behavior. Even though the above mentioned potentials for quantum correction differ in their method of calculation and their basic assumptions, yet when it comes to their inclusion into Monte Carlo simulation they are all incorporated the same way.
Simulation tool
MOCA is a full-band Monte Carlo Simulator code [15] which is suitable for 2D simulation of silicon devices at Nanohub.org. Quantum correction approach to account for size quantization in narrow channels has been adopted. Below figure shows results of sheet charges and carrier concentration inside a channel of a SOI Device at different gate biases (0, 0.25, 0.5, 0.75, and 1V).

See also
- Monte Carlo method
- Semiconductor device
- Monte Carlo method for photon transport
- Band structure
- Quantum Monte Carlo
- Quasi-Monte Carlo method
References
- ^ K. Hess, Monte Carlo Device Simulation: Full Band and Beyond, Technology (1991)
- ^ S. M. Sze. Physics of Semiconductor Devices. John Wiley and Sons, Inc (1981)
- ^ W.S. Choi, J.-K. Ahn, Y.-J. Park, H.-S. Min, and C.-G. Hwang., “A time dependent hydrodynamic device simula- tor snu-2d with new discretization scheme and algorithm.,” IEEE Trans. on CAD, vol. 13, pp. 898 (1994)
- ^ A. Forghieri, R. Guerrieri, P. Ciampolini, A. Gnudi, M. Rudan, and G. Baccarani., “A new discretization strategy of the semiconductor equations comprising momentum and energy balance,” IEEE Trans. on CAD, vol.7, pp. 231 (1988)
- ^ G. A. Sai-Halasz, M. R. Wordeman, D. P. Kern, S. Rishton, and E. Ganin, “High transconductance and velocity overshoot in NMOS devices at the 0.1 μ gate-length level,” IEEE Electron Device Letter, vol. 9, pp. 464-66 (1998)
- ^ J.H. Song, Y.J. Park, and H.S. Min, “Drain current enhancement due to velocity overshoot effects and its analytic modeling,” IEEE Trans. Electron Devices, 43, pp. 1870-5 (1996)
- ^ http://www.iue.tuwien.ac.at/phd/wessner/node31.html
- ^ Marvin L. Cohen, T. K. Bergstresser, “Band Structures and Pseudopotential Form Factors for Fourteen Semiconductors of the Diamond and Zinc-blende Structures”, Phys. Rev., vol. 141, pp. 789–796 (1966)
- ^ a b c d C. Jacoboni, L. Reggiani, “The Monte Carlo Method for Solution of Charge Transport in Semiconductor with Application to Covalent Materials” Rev. Modern Physics, vol.55, 3, pp. 645–705 (1983)
- ^ http://www.iue.tuwien.ac.at/phd/smirnov/node55.html
- ^ R. Hockney, J. Eastwood, “Computer Simulations Using Particles” McGraw Hill, Ch. 10 (1981)
- ^ D.K. Ferry, “Quantum Mechanics: An Introduction for Device Physicist and Electrical Engineer” Institute of Physics,ed. 1, p.186 (1995)
- ^ K. Hess, “Advanced Theory of Semiconductor Devices” Wiley,ed. 1, pp.94–95 (1999)
- ^ K. Hess, “Advanced Theory of Semiconductor Devices” Wiley, ed. 1, pp.97–99(1999)
- ^ https://nanohub.org/resources/moca
External links
- Nano Hub
- Illinois Tools: MOCA
- Bulk GaAs Ensemble Monte Carlo
- ECE656 Lecture31: Monte Carlo Simulation
- Quantum Corrections for Monte Carlo Simulation
- Ensemble Monte Carlo Method Described
Categories:- Monte Carlo methods
- Quantum mechanics
- Semiconductor analysis
![\frac{\partial f}{\partial t}
+ \frac{1}{\hbar} \nabla_k E(k) \nabla_r f
+ \frac{qF(r)}{\hbar} \nabla_k f
= \left[\frac{\partial f}{\partial t}\right]_\mathrm{collision}](8/81822a7e48df2d08a90fab2a944354d2.png)









![p(t) \, dt = P[k(t)] \exp[-\int^t_0 P[k(t')] \, dt' ] \, dt](e/80eccb407ec04912c1cff42fed1eb091.png)

















Wikimedia Foundation. 2010.