- SDS-PAGE
-
"PAGE" redirects here. For other uses, see Page (disambiguation).
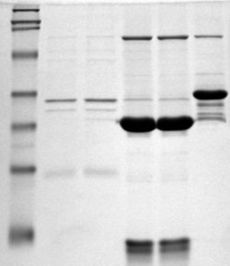 Picture of an SDS-PAGE. The molecular marker is in the left lane
Picture of an SDS-PAGE. The molecular marker is in the left lane
SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis, describes a collection of related techniques widely used in biochemistry, forensics, genetics and molecular biology to separate proteins according to their electrophoretic mobility (a function of the length of a polypeptide chain and its charge). In most proteins, the binding of SDS to the polypeptide chain imparts an even distribution of charge per unit mass, thereby resulting in a fractionation by approximate size during electrophoresis.
Contents
Procedure
Sample preparation
Samples may be any material containing proteins, such as for example prokaryotic or eukaryotic cells, tissues, viruses, environmental samples, or purified proteins. In the case of solid tissues, these are often first broken down mechanically using a blender (for larger sample volumes), using a homogenizer (smaller volumes), by sonicator or by using cycling of high pressure. Cells may also be broken open by one of the above mechanical methods.
In the case of tissues or cells, a combination of biochemical and mechanical techniques – including various types of filtration and centrifugation – may be used to separate different cell compartments and organelles prior to electrophoresis.
The sample to be analyzed is mixed with SDS, an anionic detergent which denatures secondary and non–disulfide–linked tertiary structures, and applies a negative charge to each protein in proportion to its mass. Heating the samples to at least 60 degrees C further promotes protein denaturation, helping SDS to bind.[1] [2] [3] [4]
A tracking dye may be added to the protein solution. This typically has a higher electrophoretic mobility than the proteins to allow the experimenter to track the progress of the protein solution through the gel during the electrophoretic run.

Preparing acrylamide gels
The gels typically consist of acrylamide, bisacrylamide, SDS, and a buffer with an adjusted pH. The solution may be degassed under a vacuum to prevent the formation of air bubbles during polymerization. [5] A source of free radicals and a stabilizer such as ammonium persulfate and TEMED are added to initiate polymerization.[6] The polymerization reaction results in a gel because of the added bisacrylamide, generally about 1 part in 35 relative to acrylamide, which can form cross-links between two polyacrylamide molecules. The ratio of acrylamide to bisacrylamide can be varied for special purposes. The acrylamide concentration of the gel can also be varied, generally in the range from 5% to 25%. Lower percentage gels are better for resolving very high molecular weight proteins, while much higher percentages are needed to resolve smaller proteins. Determining how much of the various solutions to mix together to make gels of particular acrylamide concentration is possible on line.

Gels are usually polymerized between two glass plates in a gel caster, with a comb inserted at the top to create the sample wells. After the gel is polymerized the comb can be removed and the gel is ready for electrophoresis.

Electrophoresis
Various buffer systems are used in SDS-PAGE depending on the nature of the sample and the experimental objective. The buffers used at the anode and cathode may be the same or different.[7][8] [9]


An electric field is applied across the gel, causing the negatively-charged proteins to migrate across the gel towards the positive (+) electrode (anode). Depending on their size, each protein will move differently through the gel matrix: short proteins will more easily fit through the pores in the gel, while larger ones will have more difficulty (they encounter more resistance). After a set amount of time (usually a few hours- though this depends on the voltage applied across the gel; higher voltages run faster but tend to produce somewhat poorer resolution), the proteins will have differentially migrated based on their size; smaller proteins will have traveled farther down the gel, while larger ones will have remained closer to the point of origin. Proteins may therefore be separated roughly according to size (and thus molecular weight); however certain glycoproteins behave anomalously on SDS gels.
Further processing
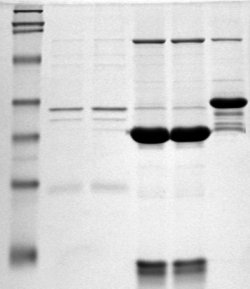
 Two SDS-PAGE-gels after a completed run
Two SDS-PAGE-gels after a completed runFollowing electrophoresis, the gel may be stained (most commonly with Coomassie Brilliant Blue R-250 or silver stain), allowing visualization of the separated proteins, or processed further (e.g. Western blot). After staining, different proteins will appear as distinct bands within the gel. It is common to run molecular weight size markers of known molecular weight in a separate lane in the gel, in order to calibrate the gel and determine the approximate molecular mass of unknown proteins by comparing the distance travelled relative to the marker.
SDS-PAGE is usually the first choice as an assay of protein purity due to its reliability and ease. The presence of SDS and the denaturing step causes proteins to be separated approximately based on size, but aberrant migration of some proteins may occur. Different proteins may also stain differently, which interferes with quantification by staining. SDS-PAGE may also be used as a preparative technique for the purification of proteins. For example, quantitative preparative native continuous polyacrylamide gel electrophoresis (QPNC-PAGE) is a method for separating native metalloproteins in complex biological matrices.
Chemical ingredients and their roles
Polyacrylamide gel (PAG) had been known as a potential embedding medium for sectioning tissues as early as 1964, and two independent groups employed PAG in electrophoresis in 1959.[10][11] It possesses several electrophoretically desirable features that make it a versatile medium. It is a synthetic, thermo-stable, transparent, strong, chemically relatively inert gel, and can be prepared with a wide range of average pore sizes.[12] The pore size of a gel is determined by two factors, the total amount of acrylamide present (%T) (T = Total concentration of acrylamide and bisacrylamide monomer) and the amount of cross-linker (%C) (C = bisacrylamide concentration). Pore size decreases with increasing %T; with cross-linking, 5%C gives the smallest pore size. Any increase or decrease in %C from 5% increases the pore size, as pore size with respect to %C is a parabolic function with vertex as 5%C. This appears to be because of non-homogeneous bundling of polymer strands within the gel. This gel material can also withstand high voltage gradients, is amenable to various staining and destaining procedures, and can be digested to extract separated fractions or dried for autoradiography and permanent recording.
 Transmission-Electron Microscopic image of a polyacrylamide gel. A polyacrylamide gel is a labyrinth of tunnels, the pore size is determined by the total amount of monomer present (%T) and the amount of cross-linker (%C).
Transmission-Electron Microscopic image of a polyacrylamide gel. A polyacrylamide gel is a labyrinth of tunnels, the pore size is determined by the total amount of monomer present (%T) and the amount of cross-linker (%C).Components
- Chemical buffer Stabilizes the pH value to the desired value within the gel itself and in the electrophoresis buffer. The choice of buffer also affects the electrophoretic mobility of the buffer counterions and thereby the resolution of the gel. The buffer should also be unreactive and not modify of react with most proteins. Different buffers may be used as cathode and anode buffers, respectively, depending on the application. Multiple pH values may be used within a single gel, for example in DISC electrophoresis. Common buffers in SDS-PAGE include Tris, Bis-Tris, or imidazole.
- Counterion balance the intrinsic charge of the buffer ion and also affect the electric field strength during electrophoresis. Highly charged and mobile ions are often avoided in SDS-PAGE cathode buffers, but may be included in the gel itself, where it migrates ahead of the protein. In applications such as DISC SDS-PAGE the pH values within the gel may vary to change the average charge of the counterions during the run to improve resolution. Popular counterions are glycine and tricine. Glycine has been used as the source of trailing ion or slow ion because its pKa is 9.69 and mobility of glycinate are such that the effective mobility can be set at a value below that of the slowest known proteins of net negative charge in the pH range. The minimum pH of this range is approximately 8.0.[
- Acrylamide (C3H5NO; mW: 71.08). When dissolved in water, slow, spontaneous autopolymerization of acrylamide takes place,joining molecules together by head on tail fashion to form long single-chain polymers. The presence of a free radical-generating system greatly accelerates polymerization. This kind of reaction is known as Vinyl addition polymerisation. A solution of these polymer chains becomes viscous but does not form a gel, because the chains simply slide over one another. Gel formation requires linking various chains together. Acrylamide is a neurotoxin. It is also essential to store acrylamide in a cool dark and dry place to reduce autopolymerisation and hydrolysis.
- Bisacrylamide (N,N'-Methylenebisacrylamide) (C7H10N2O2; mW: 154.17). Bisacrylamide is the most frequently used cross linking agent for polyacrylamide gels. Chemically it can be thought of as two acrylamide molecules coupled head to head at their non-reactive ends. Bisacrylamide can crosslink two polyacrylamide chains to one another, thereby resulting in a gel.
- Sodium Dodecyl Sulfate (SDS) (C12H25NaO4S; mW: 288.38). SDS is a strong detergent agent used to denature native proteins to unfolded, individual polypeptides. When a protein mixture is heated to 100 °C in presence of SDS, the detergent wraps around the polypeptide backbone. It binds to polypeptides in a constant weight ratio of 1.4 g SDS/g of polypeptide. In this process, the intrinsic charges of polypeptides becomes negligible when compared to the negative charges contributed by SDS. Thus polypeptides after treatment become rod-like structures possessing a uniform charge density, that is same net negative charge per unit length. The electrophoretic mobilities of these proteins will be a linear function of the logarithms of their molecular weights.
- Without SDS, different proteins with similar molecular weights would migrate differently due to differences in mass-charge ratio, as each protein has an isoelectric point and molecular weight particular to its primary structure. This is known as Native PAGE. Adding SDS solves this problem, as it binds to and unfolds the protein, giving a near uniform negative charge along the length of the polypeptide.
- Ammonium persulfate (APS) (N2H8S2O8; mW: 228.2). APS is a source of free radicals and is often used as an initiator for gel formation. An alternative source of free radicals is riboflavin, which generated free radicals in a photochemical reaction.
- TEMED (N, N, N', N'-tetramethylethylenediamine) (C6H16N2; mW: 116.21). TEMED stabilizes free radicals and improves polymerization. The rate of polymerisation and the properties of the resulting gel depend on the concentrations of free radicals. Increasing the amount of free radicals results in a decrease in the average polymer chain length, an increase in gel turbidity and a decrease in gel elasticity. Decreasing the amount shows the reverse effect. The lowest catalytic concentrations that will allow polymerisation in a reasonable period of time should be used. APS and TEMED are typically used at approximately equimolar concentrations in the range of 1 to 10 mM.
Chemicals for processing and visualization
The following chemicals are used for processing of the gel and the protein samples visualized in it:
- Tracking dye. As proteins are mostly colourless, their progress through the gel during electrophoresis cannot be easily followed. Anionic dyes of a known electrophoretic mobility are therefore usually included in the SDS-PAGE sample buffer. A very common tracking dye is Bromophenol blue (BPB, 3',3",5',5" tetrabromophenolsulfonphthalein). This dye is coloured at alkali and neutral pH and is a small negatively charged molecule that moves towards the anode. Being a highly mobile molecule it moves ahead of most proteins. As it reaches the anodic end of the electrophoresis medium electrophoresis is stopped. It can weakly bind to some proteins and impart a blue colour.
- Loading aids. Most SDS-PAGE systems are loaded from the top into wells within the gel. To ensure that the sample sinks to the bottom of the gel, sample buffer is supplemented with additives that increase the density of the sample. These additives should be non-ionic and non-reactive towards proteins to avoid interfering with electrophoresis. Common additives are glycerol and sucrose.
- Coomassie Brilliant Blue R-250 (CBB)(C45H44N3NaO7S2; mW: 825.97). CBB is the most popular protein stain. It is an anionic dye, which non-specifically binds to proteins. The structure of CBB is predominantly non-polar, and it is usually used in methanolic solution acidified with acetic acid. Proteins in the gel are fixed by acetic acid and simultaneously stained. The excess dye incorporated into the gel can be removed by destaining with the same solution without the dye. The proteins are detected as blue bands on a clear background. As SDS is also anionic, it may interfere with staining process. Therefore, large volume of staining solution is recommended, at least ten times the volume of the gel.
Reducing SDS-PAGE
Besides the addition of SDS, proteins may optionally be briefly heated to near boiling in the presence of a reducing agent, such as dithiothreitol (DTT) or 2-mercaptoethanol (beta-mercaptoethanol/BME), which further denatures the proteins by reducing disulfide linkages, thus overcoming some forms of tertiary protein folding, and breaking up quaternary protein structure (oligomeric subunits). This is known as reducing SDS-PAGE, and is most commonly used.
Silver staining
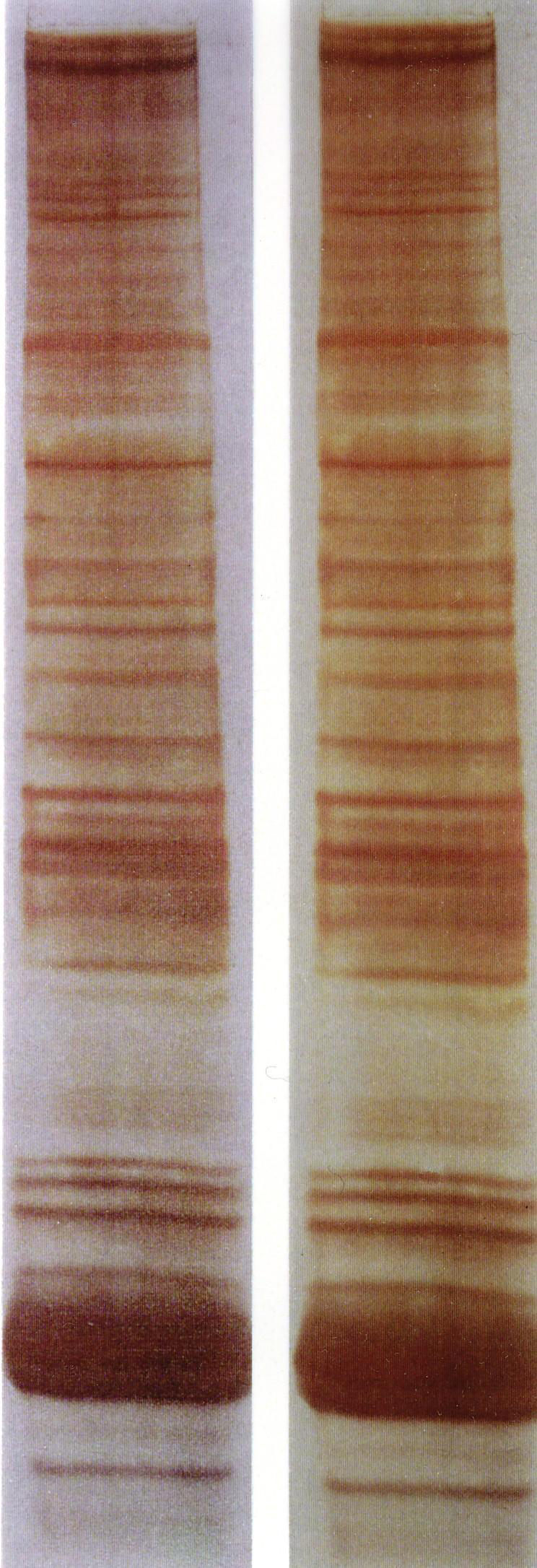
 Silver stained SDS Polyacrylamide gels
Silver stained SDS Polyacrylamide gelsIn the 14th century the silver staining technique was developed for colouring the surface of glass. It has been used extensively for this purpose since the 16th century. The colour produced by the early silver stains ranged between light yellow and an orange-red. Camillo Golgi perfected the silver staining for the study of the nervous system. Golgi's method stains a limited number of cells at random in their entirety.[13] The exact chemical mechanism by which this happens is still largely unknown.[14] Silver staining was introduced by Kerenyi and Gallyas as a sensitive procedure to detect trace amounts of proteins in gels.[15] The technique has been extended to the study of other biological macromolecules that have been separated in a variety of supports.[16] Classical Coomassie Brilliant Blue staining can usually detect a 50 ng protein band, Silver staining increases the sensitivity typically 50 times. Many variables can influence the colour intensity and every protein has its own staining characteristics; clean glassware, pure reagents and water of highest purity are the key points to successful staining.[17]
Buffer systems
 Postulated migration of proteins in a Laemmli gel system A: Stacking gel, B: Resolving gel, o: sample application c: discontinuities in the buffer and electrophoretic matrix
Postulated migration of proteins in a Laemmli gel system A: Stacking gel, B: Resolving gel, o: sample application c: discontinuities in the buffer and electrophoretic matrixMost protein separations are performed using a "discontinuous" (or DISC) buffer system that significantly enhances the sharpness of the bands within the gel. During electrophoresis in a discontinuous gel system, an ion gradient is formed in the early stage of electrophoresis that causes all of the proteins to focus into a single sharp band. The formation of the ion gradient is achieved by choosing a pH value at which the ions of the buffer are only moderately charged compared to the SDS-coated proteins. These conditions provide an environment in which Kohlrausch reactions determine the molar conductivity. As a result, SDS-coated proteins are concentrated to several fold in a thin zone of the order of 19 μm within a few minutes. At this stage all proteins migrate at the same migration speed by isotachophoresis. This occurs in a region of the gel that has larger pores so that the gel matrix does not retard the migration during the focusing or "stacking" event. [18][19] Separation of the proteins by size is achieved in the lower, "resolving" region of the gel. The resolving gel typically has a much smaller pore size, which leads to a sieving effect that now determines the electrophoretic mobility of the proteins. At the same time, the separating part of the gel also has an pH value in which the buffer ions on average carry a greater charge, causing them to "outrun" the SDS-covered proteins and eliminate the ion gradient and thereby the stacking effect.
A very widespread discontinuous buffer system is the tris-glycine or "Laemmli" system that stacks at a pH of 6.8 and resolves at a pH of ~8.3-9.0. A drawback of this system is that these pH values may promote disulfide bond formation between cysteine residues in the proteins because the pKa of cysteine ranges from 8-9 and because reducing agent present in the loading buffer doesn't co-migrate with the proteins. Recent advances in buffering technology alleviate this problem by resolving the proteins at a pH well below the pKa of cysteine (e.g., bis-tris, pH 6.5) and include reducing agents (e.g. sodium bisulfite) that move into the gel ahead of the proteins to maintain a reducing environment. An additional benefit of using buffers with lower pH values is that the acrylamide gel is more stable lower pH values, so the gels can be stored for long periods of time before use.[20][21]
SDS gradient gel electrophoresis of proteins
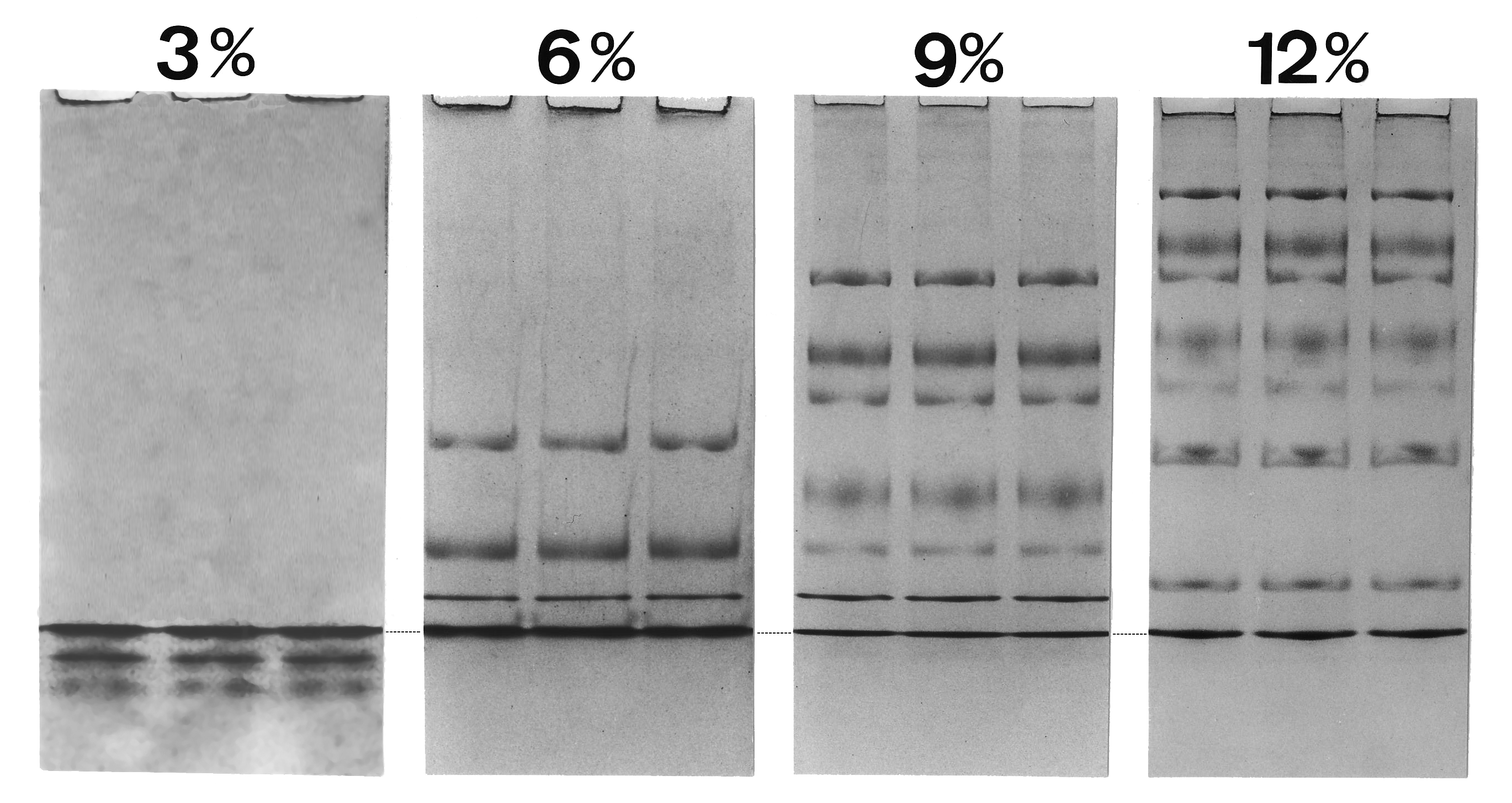
 Migration of proteins in SDS gels of varying acrylamide concentrations (%T). The migration of nine proteins ranging from 94 kDa to 14.4 kDa is shown. Stacking and unstacking occurs continuously in the gel, for every protein at a different gel concentration. The dotted line indicates the discontinuity at the Gly¯/Cl¯ moving boundary. Proteins between the fast leading electrolyte and the slow trailing electrolyte are not diluted by diffusion.
Migration of proteins in SDS gels of varying acrylamide concentrations (%T). The migration of nine proteins ranging from 94 kDa to 14.4 kDa is shown. Stacking and unstacking occurs continuously in the gel, for every protein at a different gel concentration. The dotted line indicates the discontinuity at the Gly¯/Cl¯ moving boundary. Proteins between the fast leading electrolyte and the slow trailing electrolyte are not diluted by diffusion.As voltage is applied, the anions (and negatively charged sample molecules) migrate toward the positive electrode (anode) in the lower chamber, the leading ion is Cl¯ ( high mobility and high concentration); glycinate is the trailing ion (low mobility and low concentration). SDS-protein particles do not migrate freely at the border between the Cl¯ of the gel buffer and the Gly¯ of the cathode buffer. Friedrich Kohlrausch found that Ohm's law also applies to dissolved electrolytes. Because of the voltage drop between the Cl- and Glycine-buffers, proteins are compressed (stacked) into micrometer thin layers.[22] The boundary moves through a pore gradient and the protein stack gradually disperses due to a frictional resistance increase of the gel matrix. Stacking and unstacking occurs continuously in the gradient gel, for every protein at a different position. For a complete protein unstacking the polyacrylamide-gel concentration must exceed 16% T. The two-gel system of "Laemmli" is a simple gradient gel. The pH discontinuity of the buffers is of no significance for the separation quality, and a "stacking-gel" with a different pH is not needed.
See also
- Capillary electrophoresis
- DNA electrophoresis
- Eastern blotting
- Electroblotting
- Electrophoresis
- Gel electrophoresis
- History of electrophoresis
- Isoelectric focusing
- Isotachophoresis
- Native Gel Electrophoresis
- Northern blotting
- Protein electrophoresis
- Southern blotting
- Two dimensional SDS-PAGE
- Western blotting
- Zymography
References
- ^ Shapiro AL, Viñuela E, Maizel JV Jr. (September 1967). "Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels.". Biochem Biophys Res Commun. 28 (5): 815–820. doi:10.1016/0006-291X(67)90391-9. PMID 4861258.
- ^ Weber K, Osborn M (August 1969). "The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis.". J Biol Chem. 244 (16): 4406–4412. PMID 5806584.
- ^ Laemmli UK (August 1970). "Cleavage of structural proteins during the assembly of the head of bacteriophage T4". Nature 227 (5259): 680–685. doi:10.1038/227680a0. PMID 5432063.
- ^ Caprette, David. "SDS-PAGE". http://www.ruf.rice.edu/~bioslabs/studies/sds-page/denature.html. Retrieved 27 September 2009.
- ^ "What is the meaning of de -gas the acrylamide gel mix?". http://www.protocol-online.org/biology-forums/posts/17740.html. Retrieved 28 September 2009.
- ^ "SDS-PAGE". http://www.science.smith.edu/departments/Biochem/Biochem_353/sdspage.html. Retrieved 12 September 2009.
- ^ Laemmli UK (August 1970). "Cleavage of structural proteins during the assembly of the head of bacteriophage T4". Nature 227 (5259): 680–685. doi:10.1038/227680a0. PMID 5432063.
- ^ Schägger H, von Jagow G (Nov 1987). "Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa". Anal Biochem 166 (2): 368–379. doi:10.1016/0003-2697(87)90587-2. PMID 2449095.
- ^ Andrews. "SDS-PAGE". http://www.dwalab.ca/labman/recipes/PreparingtoRunTricineSDSGels.html. Retrieved 27 September 2009.
- ^ Davis BJ, Ornstein L (1959). "A new high resolution electrophoresis method.". Delivered at the Society for the Study of Blood at the New York Academy of Medicine.
- ^ Raymond S, Weintraub L. (1959). "Acrylamide gel as a supporting medium for zone electrophoresis.". Science 130 (3377): 711. doi:10.1126/science.130.3377.711. PMID 14436634.
- ^ Rüchel R, Steere RL, Erbe EF (1978). "Transmission-electron microscopic observations of freeze-etched polyacrylamide gels". J Chromatogr. 166 (2): 563–575. doi:10.1016/S0021-9673(00)95641-3.
- ^ Grant G (Oct 2007). "How the 1906 Nobel Prize in Physiology or Medicine was shared between Golgi and Cajal". Brain Res Rev 55 (2): 490–498. doi:10.1016/j.brainresrev.2006.11.004. PMID 17306375.
- ^ Golgi C (1873). "Sulla struttura della sostanza grigia del cervello". Gazzetta Medica Italiana (Lombardia) 33: 244–246.
- ^ Kerenyi L, Gallyas F (1973). "Über Probleme der quantitiven Auswertung der mit physikalischer Entwicklung versilberten Agarelektrophoretogramme". Clin. Chim. Acta 47 (3): 425–436. doi:10.1016/0009-8981(73)90276-3. PMID 4744834.
- ^ Switzer RC 3rd, Merril CR, Shifrin S (Sep 1979). "A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels". Anal Biochem. 98 (1): 231–237. doi:10.1016/0003-2697(79)90732-2. PMID 94518.
- ^ Hempelmann E, Schulze M, Götze O (1984). "Free SH-groups are important for the polychromatic staining of proteins with silver nitrat". Neuhof V (ed)Electrophoresis '84 , Verlag Chemie Weinheim 1984: 328–330.
- ^ Ornstein L (December 1964). "DISC ELECTROPHORESIS. I. BACKGROUND AND THEORY". Ann N Y Acad Sci. 121 (2): 321–349. doi:10.1111/j.1749-6632.1964.tb14207.x. PMID 14240533.
- ^ Davis BJ (December 1964). "Disc Electrophoresis. 2, Method and application to human serum proteins". Ann. New York Acad. Sci 121 (2): 404–427. doi:10.1111/j.1749-6632.1964.tb14213.x. PMID 14240539.
- ^ Schägger H, von Jagow G (1987). "Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa". Anal Biochem. 166 (2): 368–379. doi:10.1016/0003-2697(87)90587-2. PMID 2449095.
- ^ Wiltfang J, Arold N, Neuhoff V (1991). "A new multiphasic buffer system for sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100,000-1000, and their detection with picomolar sensitivity". Electrophoresis 12 (5): 352–366. doi:10.1002/elps.1150120507. PMID 1718736.
- ^ Kohlrausch F (1897). "Ueber Concentrations-Verschiebungen durch Electrolyse im Inneren von Lösungen und Lösungsgemischen". Ann.J.Phys.u.Chem. 62 (10): 209–239. doi:10.1002/andp.18972981002.
External links
- Demystifying SDS-PAGE Video
- Demystifying SDS-PAGE
- SDS-PAGE Calculator for customised recipes for TRIS Urea gels.
- 2-Dimensional Protein Gelelectrophoresis
- [1] Hempelmann E. SDS-Protein PAGE and Proteindetection by Silverstaining and Immunoblotting of Plasmodium falciparum proteins. in: Moll K, Ljungström J, Perlmann H, Scherf A, Wahlgren M (eds) Methods in Malaria Research, 5th edition, 2008, 263-266
Proteins: key methods of study Experimental Protein purification · Green fluorescent protein · Western blot · Protein immunostaining · Protein sequencing · Gel electrophoresis/Protein electrophoresis · Protein immunoprecipitation · Peptide mass fingerprinting · Dual polarization interferometry · Microscale thermophoresis · Chromatin immunoprecipitationBioinformatics Protein structure prediction · Protein–protein docking · Protein structural alignment · Protein ontology · Protein–protein interaction predictionAssay Display techniques Super resolution microscopy Vertico SMICategories:



Wikimedia Foundation. 2010.