- Enthalpy
-
Enthalpy is a measure of the total energy of a thermodynamic system. It includes the internal energy, which is the energy required to create a system, and the amount of energy required to make room for it by displacing its environment and establishing its volume and pressure.
Enthalpy is a thermodynamic potential. It is a state function and an extensive quantity. The unit of measurement for enthalpy in the International System of Units (SI) is the joule, but other historical, conventional units are still in use, such as the British thermal unit and the calorie.
The enthalpy is the preferred expression of system energy changes in many chemical, biological, and physical measurements, because it simplifies certain descriptions of energy transfer. This is because a change in enthalpy takes account of energy transferred to the environment through the expansion of the system under study.
The total enthalpy, H, of a system cannot be measured directly. Thus, change in enthalpy, ΔH, is a more useful quantity than its absolute value. The change ΔH is positive in endothermic reactions, and negative in exothermic processes. ΔH of a system is equal to the sum of non-mechanical work done on it and the heat supplied to it.
For quasistatic processes under constant pressure, ΔH is equal to the change in the internal energy of the system, plus the work that the system has done on its surroundings.[1] This means that the change in enthalpy under such conditions is the heat absorbed (or released) by a chemical reaction.
Contents
Formal definition
H(S,p), can be derived as a thermodynamic potential naturally dependent on S and p can be derived as follows from U(S,V). Here, U is internal energy, T is absolute temperature, S is entropy, p is pressure, and V is volume: The fundamental thermodynamic relation is basically the First Law of thermodynamics for reversible processes:
- dU = TdS − pdV
Apply the product for differentiation to pV:
- dU = TdS + Vdp − d(pV), hence
- d(U + pV) = TdS + Vdp
The enthalpy of a system is defined as:
so
- dH(S,p) = TdS + Vdp
where
- H is the enthalpy of the system
- U is the internal energy of the system
- p is the pressure at the boundary of the system and its environment
- V is the volume of the system.
Note that the U term is equivalent to the energy required to create the system, and that the pV term is equivalent to the energy that would be required to "make room" for the system if the pressure of the environment remained constant.
The pV term may be understood by the following example of an isobaric process. Consider gas changing its volume (by, for example, a chemical reaction) in a cylinder, pushing a piston, maintaining constant pressure p. The force is calculated from the area A of the piston and definition of pressure p = F/A: the force is F = pA. By definition, work W done is W = Fx, where x is the distance traversed. Combining gives W = pAx, and the product Ax is the volume traversed by the piston: Ax = V. Thus, the work done by the gas is W = pV, where p is a constant pressure and V the expansion of volume. Including this pV term means that during constant pressure expansion, any internal energy forfeited as work on the environment does not affect the value of enthalpy. The enthalpy change can be defined ΔH = ΔU + W = ΔU + Δ(pV), where ΔU is the thermal energy lost to expansion, and W the energy gained due to work done on the piston.
Difference between enthalpy and internal energy
Chemists routinely use H as the energy of the system, but the pV term is not stored in the system, but rather in the surroundings, such as the atmosphere. When a system, for example, n moles of a gas of volume V at pressure P and temperature T, is created or brought to its present state from absolute zero, energy must be supplied equal to its internal energy U plus pV, where pV is the work done in pushing against the ambient (atmospheric) pressure. This additional energy is, therefore, stored in the surroundings and can be recovered when the system collapses back to its initial state. In basic chemistry scientists are typically interested in experiments conducted at atmospheric pressure, and for reaction energy calculations they care about the total energy in such conditions, and therefore typically need to use H. In basic physics and thermodynamics it may be more interesting to study the internal properties of the system and therefore the internal energy is used.[citation needed]
Origins
The word enthalpy is based on the Greek word enthalpos (ἔνθαλπος), which means to put heat into. It comes from the Classical Greek prefix ἐν-, en-, meaning to put into, and the verb θάλπειν, thalpein, meaning "to heat". The word enthalpy is often incorrectly attributed[citation needed] to Benoit Paul Émile Clapeyron and Rudolf Clausius through the 1850 publication of their Clausius-Clapeyron relation. This misconception was popularized by the 1927 publication of The Mollier Steam Tables and Diagrams. However, neither the concept, the word, nor the symbol for enthalpy existed until well after Clapeyron's death.
The earliest writings to contain the concept of enthalpy did not occur until 1875,[2] when Josiah Willard Gibbs introduced "a heat function for constant pressure". However, Gibbs did not use the word "enthalpy" in his writings.[note 1] Instead, the word "enthalpy" first appears in the scientific literature in a 1909 publication by J. P. Dalton. According to that publication, the word was actually coined by Heike Kamerlingh Onnes.[3] Over the years, many different symbols were used to denote enthalpy. It was not until 1922 that Alfred W. Porter proposed the symbol "H" as the accepted standard,[4] thus finalizing the terminology still in use today.
Relationship to heat
The increase in enthalpy of a system is exactly equal to the energy added through heat, provided that the system is under constant pressure and that the only work done on the system is expansion work:
where
- ΔH is the change in enthalpy of the system (under the restrictions mentioned above), and
- Q is the energy added to the system through heat.
Expansion work is the transfer of energy between the system and its environment through changes in the system's volume. This type of work does not affect the above equation. Any other type of work that could be done on the system aside from expansion is called non-mechanical work.[5] Non-mechanical work could include such processes as altering the internal energy using an external electric field, or adding energy through stirring. If any non-mechanical work takes place then the above equation will not hold.
The exact relationship between enthalpy and heat can be derived from the definition of enthalpy.
According to the definition of enthalpy,
where
- H is the enthalpy of the system,
- U is the internal energy of the system,
- p is the pressure at the boundary of the system and its environment, and
- V is the volume of the system.
Differentiating yields
According to the first law of thermodynamics, any changes in internal energy are due to energy transferred with the environment (dU = δQ + δW + δW'), so
where
- δQ is the infinitesimal amount of energy added to the system through heat
- δW is the infinitesimal amount of energy added to the system through expansion work
- δW' is the infinitesimal amount of energy added to the system through any means other than heat or expansion work. (W' is sometimes called non-mechanical work.[5])
- (note that the inexact differential, δ, is required for the path-dependent variables Q, W, and W')
Because the energy added to the system through expansion work is δW = − pdV, this term can cancel with the existing pdV term to yield:
integrating then yields
Notice that this equation still has some extra terms; this is where the restrictions come in. Restricting the conditions to constant pressure ensures that the final term will equal zero. Ensuring that no work is done aside from expansion work makes the W' term zero. Thus
Provided that the pressure is constant and that the only work done on the system is through system expansion.
Enthalpy is not heat
Enthalpy is sometimes described as the heat content of a system under a given pressure, whereas "heat" is defined as thermal energy in transit. For the assumption that a change of enthalpy ΔH is heat to be valid, no energy exchange with the environment must occur aside from heat or expansion work. Given this restriction, it can be shown that:
- The enthalpy is the total amount of energy that the system can emit through heat
- Adding or removing energy through heat is the only way to change the enthalpy
- The amount of change in enthalpy is equal to the amount of energy added through heat.
Thus it is as if enthalpy is nothing more than heat "stored" by the system, provided the given restrictions are adhered to.
However, heat is not the only way to change enthalpy. Enthalpy also changes when the pressure of the environment is altered, even if no energy is exchanged as heat. In addition, enthalpy changes when energy is transferred into or out of the system through a means other than heat or expansion work, such as through external fields or stirring.
Applications
In thermodynamics, one can calculate enthalpy by determining the requirements for creating a system from "nothingness"; the mechanical work required, pV, differs based upon the constancy of conditions present at the creation of the thermodynamic system.
Internal energy, U, must be supplied to remove particles from a surrounding in order to allow space for the creation of a system, providing that environmental variables, such as pressure (p) remain constant. This internal energy also includes the energy required for activation and the breaking of bonded compounds into gaseous species.
This process is calculated within enthalpy calculations as U + pV, to label the amount of energy or work required to "set aside space for" and "create" the system; describing the work done by both the reaction or formation of systems, and the surroundings. For systems at constant pressure, the change in enthalpy is the heat received by the system.
Therefore, the change in enthalpy can be devised or represented without the need for compressive or expansive mechanics; for a simple system, with a constant number of particles, the difference in enthalpy is the maximum amount of thermal energy derivable from a thermodynamic process in which the pressure is held constant.
The term pV is the work required to displace the surrounding atmosphere in order to vacate the space to be occupied by the system.
Heat of reaction
The total enthalpy of a system cannot be measured directly; the enthalpy change of a system is measured instead. Enthalpy change is defined by the following equation:
- ΔH = Hfinal − Hinitial
where
- ΔH is the enthalpy change
- Hfinal is the final enthalpy of the system, expressed in joules. In a chemical reaction, Hfinal is the enthalpy of the products.
- Hinitial is the initial enthalpy of the system, expressed in joules. In a chemical reaction, Hinitial is the enthalpy of the reactants.
For an exothermic reaction at constant pressure, the system's change in enthalpy equals the energy released in the reaction, including the energy retained in the system and lost through expansion against its surroundings. In a similar manner, for an endothermic reaction, the system's change in enthalpy is equal to the energy absorbed in the reaction, including the energy lost by the system and gained from compression from its surroundings. A relatively easy way to determine whether or not a reaction is exothermic or endothermic is to determine the sign of ΔH. If ΔH is positive, the reaction is endothermic, that is heat is absorbed by the system due to the products of the reaction having a greater enthalpy than the reactants. On the other hand if ΔH is negative, the reaction is exothermic, that is the overall decrease in enthalpy is achieved by the generation of heat.
Although enthalpy is commonly used in engineering and science, it is impossible to measure directly, as enthalpy has no datum (reference point). Therefore enthalpy can only accurately be used in a closed system. However, few real-world applications exist in closed isolation, and it is for this reason that two or more closed systems cannot be compared using enthalpy as a basis, although sometimes this is done erroneously.
Specific enthalpy
The specific enthalpy of a working mass is a property of that mass used in thermodynamics. It is defined as h = u + pv, where u is the specific internal energy, p is the pressure, and v is specific volume. In other words, h = H/m where m is the mass of the system. The SI unit for specific enthalpy is joules per kilogram.
Enthalpy changes
An enthalpy change describes the change in enthalpy observed in the constituents of a thermodynamic system when undergoing a transformation or chemical reaction. It is the difference between the enthalpy after the process has completed, i.e. the enthalpy of the products, and the initial enthalpy of the system, i.e. the reactants. These processes are reversible and the enthalpy for the reverse process is the negative value of the forward change.
A common standard enthalpy change is the enthalpy of formation, which has been determined for a large number of substances. Enthalpy changes are routinely measured and compiled in chemical and physical reference works, such as the CRC Handbook of Chemistry and Physics. The following is a selection of enthalpy changes commonly recognized in thermodynamics.
When used in these recognized terms the qualifier change is usually dropped and the property is simply termed enthalpy of 'process'. Since these properties are often used as reference values it is very common to quote them for a standardized set of environmental parameters, or standard conditions, which is typically a temperature of 298K and a pressure of either 1atm or 101.3kPa. For such standardized values the name of the enthalpy is commonly prefixed with the term standard, e.g. standard enthalpy of formation.
Chemical properties:
- Enthalpy of reaction, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of substance reacts completely.
- Enthalpy of formation, defined as the enthalpy change observed in a constituent of a thermodynamic system when, one mole of a compound is formed from its elementary antecedents.
- Enthalpy of combustion, defined as the enthalpy change observed in a constituent of a thermodynamic system, when one mole of a substance combusts completely with oxygen.
- Enthalpy of hydrogenation, defined as the enthalpy change observed in a constituent of a thermodynamic system, when one mole of an unsaturated compound reacts completely with an excess of hydrogen to form a saturated compound.
- Enthalpy of atomization, defined as the enthalpy change required to atomize one mole of compound completely.
- Enthalpy of neutralization, defined as the enthalpy change observed in a constituent of a thermodynamic system, when one mole of water is produced when an acid and a base react.
- Standard Enthalpy of solution, defined as the enthalpy change observed in a constituent of a thermodynamic system, when one mole of an solute is dissolved completely in an excess of solvent.
- Standard enthalpy of Denaturation (biochemistry), defined as the enthalpy change required to denature one mole of compound.
- Enthalpy of hydration, defined as the enthalpy change observed when one mole of gaseous ions are completely dissolved in water forming one mole of aqueous ions.
Physical properties:
- Enthalpy of fusion, defined as the enthalpy change required to completely change the state of one mole of substance between solid and liquid states.
- Enthalpy of vaporization, defined as the enthalpy change required to completely change the state of one mole of substance between liquid and gaseous states.
- Enthalpy of sublimation, defined as the enthalpy change required to completely change the state of one mole of substance between solid and gaseous states.
- Lattice enthalpy, defined as the energy required to separate one mole of an ionic compound into separated gaseous ions to an infinite distance apart (meaning no force of attraction).
Open systems
In thermodynamic open systems, matter may flow in and out of the system boundaries. The first law of thermodynamics for open systems states: The increase in the internal energy of a system is equal to the amount of energy added to the system by matter flowing in and by heating, minus the amount lost by matter flowing out and in the form of work done by the system. The first law for open systems is given by:
- dU = dUin − dUout + δQ − δW
where Uin is the average internal energy entering the system and Uout is the average internal energy leaving the system.
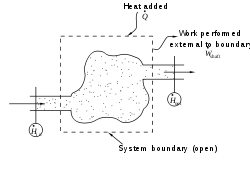 During steady, continuous operation, an energy balance applied to an open system equates shaft work performed by the system to heat added plus net enthalpy added
During steady, continuous operation, an energy balance applied to an open system equates shaft work performed by the system to heat added plus net enthalpy added
The region of space enclosed by open system boundaries is usually called a control volume, and it may or may not correspond to physical walls. If we choose the shape of the control volume such that all flow in or out occurs perpendicular to its surface, then the flow of matter into the system performs work as if it were a piston of fluid pushing mass into the system, and the system performs work on the flow of matter out as if it were driving a piston of fluid. There are then two types of work performed: flow work described above, which is performed on the fluid (this is also often called pV work), and shaft work, which may be performed on some mechanical device.
These two types of work are expressed in the equation:
- δW = d(poutVout) − d(pinVin) + δWshaft.
Substitution into the equation above for the control volume cv yields:
- dUcv = dUin + d(pinVin) − dUout − d(poutVout) + δQ − δWshaft.
The definition of enthalpy, H, permits us to use this thermodynamic potential to account for both internal energy and pV work in fluids for open systems:
- dUcv = dHin − dHout + δQ − δWshaft.
Note that the previous expression holds true only if the kinetic energy flow rate is conserved between system inlet and outlet. Otherwise, it has to be included in the enthalpy balance. During steady-state operation of a device (see turbine, pump, and engine), the expression above may be set equal to zero. This yields a useful expression for the power generation or requirement for these devices in the absence of chemical reactions:
This expression is described by the diagram above.
Other formulas
As an expansion of the first law of thermodynamics, enthalpy can be related to several other thermodynamic formulas. As with the original definition of the first law;
where, as defined by the law;
- dU represents the infinitesimal increase of the systematic or internal energy
- δQ represents the infinitesimal amount of energy attributed or added to the system
- δW represents the infinitesimal amount of energy acted out by the system on the surroundings.
As a differential expression, the value of H can be defined as[6]
where
- δ represents the inexact differential
- U is the internal energy
- δQ = TdS is the energy added by heating during a reversible process
- δW = pdV is the work done by the system in a reversible process
- dS is the increase in entropy (joules per kelvin)
- p is the constant pressure
- dV is an infinitesimal volume
- T is the temperature (kelvin).
For a process that is not reversible, the above equation expressing dH in terms of dS and dp still holds because H is a thermodynamic state variable that can be uniquely specified by S and p. We thus have in general:
- dH = TdS + Vdp
It is seen that, if a thermodynamic process is isobaric (i.e., occurs at constant pressure), then dp is zero and thus
- dH = TdS ≥ δQ
The difference in enthalpy is the maximum thermal energy attainable from the system in an isobaric process. This explains why it is sometimes called the heat content. That is, the integral of dH over any isobar in state space is the maximum thermal energy attainable from the system.
In a more general form, the first law describes the internal energy with additional terms involving the chemical potential and the number of particles of various types. The differential statement for dH then becomes:
where μi is the chemical potential for an i-type particle, and Ni is the number of such particles. It is seen that, not only must the Vdp term be set to zero by requiring the pressures of the initial and final states to be the same, but the μidNi terms must be zero as well, by requiring that the particle numbers remain unchanged. Any further generalization will add even more terms whose extensive differential term must be set to zero in order for the interpretation of the enthalpy to hold.
See also
- Standard enthalpy change of formation (data table)
- Calorimetry
- Calorimeter
- Departure function
- Entropy
- Gibbs free energy
- Hess's law
- Isenthalpic process
- Stagnation enthalpy
- Thermodynamic databases for pure substances
Notes
- ^ The Collected Works of J. Willard Gibbs, Vol. I do not contain reference to the word enthalpy, but rather reference the heat function for constant pressure.
References
- ^ G.J. Van Wylen and R.E. Sonntag (1985), Fundamentals of Classical Thermodynamics, Section 5.5 (3rd edition), John Wiley & Sons Inc. New York, NY. ISBN 0-471-82933-1
- ^ Henderson, Douglas; Eyring, Henry; Jost, Wilhelm (1967). Physical Chemistry: An Advanced Treatise. Academic Press. p. 29.
- ^ Laidler, Keith (1995). The World of Physical Chemistry. Oxford University Press. p. 110.
- ^ Howard, Irmgard (2002). "H Is for Enthalpy, Thanks to Heike Kamerlingh Onnes and Alfred W. Porter". Journal of Chemical Education (ACS Publications) 79 (6): 697. Bibcode 2002JChEd..79..697H. doi:10.1021/ed079p697. http://pubs.acs.org/doi/abs/10.1021/ed079p697.
- ^ a b Bokshteĭn, Boris; Mendelev, Mikhail; Srolovitz, David (2005). Thermodynamics and kinetics in materials science: a short course. Oxford University Press. p. 7. ISBN 9780198528043. http://books.google.com/?id=V1bqHaR0HY0C&pg=PA7&lpg=PA7&dq=%22called+non-mechanical+work%22&q=%22called%20non-mechanical%20work%22.
- ^ DeHoff p 57
Bibliography
- Haase, R. In Physical Chemistry: An Advanced Treatise; Jost, W., Ed.; Academic: New York, 1971; p 29.
- Gibbs, J. W. In The Collected Works of J. Willard Gibbs, Vol. I; Yale University Press: New Haven, CT, reprinted 1948; p 88.
- Laidler, K. The World of Physical Chemistry; Oxford University Press: Oxford, 1995; p 110.
- C.Kittel, H.Kroemer In Thermal Physics; S.R Furphy and Company, New York, 1980; p246
- DeHoff, R. Thermodynamics in Materials Science: 2nd ed.; Taylor and Francis Group, New York, 2006.
External links
- Enthalpy - Eric Weisstein's World of Physics
- Enthalpy - Georgia State University
- Enthalpy example calculations - Texas A&M University Chemistry Department
Categories:- State functions
- Enthalpy
- Fundamental physics concepts
- dU = TdS − pdV


















Wikimedia Foundation. 2010.