- Gibbs free energy
-
In thermodynamics, the Gibbs free energy (IUPAC recommended name: Gibbs energy or Gibbs function; also known as free enthalpy[1] to distinguish it from Helmholtz free energy) is a thermodynamic potential that measures the "useful" or process-initiating work obtainable from a thermodynamic system at a constant temperature and pressure (isothermal, isobaric). Just as in mechanics, where potential energy is defined as capacity to do work, similarly different potentials have different meanings. Gibbs energy is the capacity of a system to do non-mechanical work and ΔG measures the non-mechanical work done on it. The Gibbs free energy is the maximum amount of non-expansion work that can be extracted from a closed system; this maximum can be attained only in a completely reversible process. When a system changes from a well-defined initial state to a well-defined final state, the Gibbs free energy ΔG equals the work exchanged by the system with its surroundings, minus the work of the pressure forces, during a reversible transformation of the system from the same initial state to the same final state.[2]
Gibbs energy (also referred to as ∆G) is also the chemical potential that is minimized when a system reaches equilibrium at constant pressure and temperature. Its derivative with respect to the reaction coordinate of the system vanishes at the equilibrium point. As such, it is a convenient criterion of spontaneity for processes with constant pressure and temperature.
The Gibbs free energy, originally called available energy, was developed in the 1870s by the American mathematician Josiah Willard Gibbs. In 1873, Gibbs described this “available energy” as
the greatest amount of mechanical work which can be obtained from a given quantity of a certain substance in a given initial state, without increasing its total volume or allowing heat to pass to or from external bodies, except such as at the close of the processes are left in their initial condition.[3]The initial state of the body, according to Gibbs, is supposed to be such that "the body can be made to pass from it to states of dissipated energy by reversible processes." In his 1876 magnum opus On the Equilibrium of Heterogeneous Substances, a graphical analysis of multi-phase chemical systems, he engaged his thoughts on chemical free energy in full.
Contents
Overview
In a simple manner, with respect to STP reacting systems, a general rule of thumb is:
“ Every system seeks to achieve a minimum of free energy. ” Hence, out of this general natural tendency, a quantitative measure as to how near or far a potential reaction is from this minimum is when the calculated energetics of the process indicate that the change in Gibbs free energy ΔG is negative. In essence, this means that such a reaction will be favoured and will release energy. The energy released equals the maximum amount of work that can be performed as a result of the chemical reaction. In contrast, if conditions indicated a positive ΔG, then energy—in the form of work—would have to be added to the reacting system to make the reaction go.
The equation can also be seen from the perspective of both the system and its surroundings (the universe). For the purposes of calculation, we assume the reaction is the only reaction going on in the universe. Thus the entropy released or absorbed by the system is actually the entropy that the environment must absorb or release respectively. Thus the reaction will only be allowed if the total entropy change of the universe is equal to zero (an equilibrium process) or positive. The input of heat into an "endothermic" chemical reaction (e.g. the elimination of cyclohexanol to cyclohexene) can be seen as coupling an inherently unfavourable reaction (elimination) to a favourable one (burning of coal or the energy source of a heat source) such that the total entropy change of the universe is more than or equal to zero, making the Gibbs free energy of the coupled reaction negative.
In traditional use, the term “free” was attached to Gibbs free energy for systems at constant pressure and temperature to mean "available in the form of useful work."[2] For Gibbs free energy, we add the qualification that it is the energy free for non-volume work.[4] (A similar meaning applies used in conjunction with Helmholtz free energy, for systems at constant volume and temperature). However, an increasing number of books and journal articles do not include the attachment “free”, referring to G as simply "Gibbs energy". This is the result of a 1988 IUPAC meeting to set unified terminologies for the international scientific community, in which the adjective ‘free’ was supposedly banished.[5] This standard, however, has not yet been universally adopted.
History
See also: Thermodynamic free energyThe quantity called "free energy" is a more advanced and accurate replacement for the outdated term affinity, which was used by chemists in previous years to describe the force that caused chemical reactions.
In 1873, Willard Gibbs published A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces, in which he introduced the preliminary outline of the principles of his new equation able to predict or estimate the tendencies of various natural processes to ensue when bodies or systems are brought into contact. By studying the interactions of homogeneous substances in contact, i.e., bodies, being in composition part solid, part liquid, and part vapor, and by using a three-dimensional volume-entropy-internal energy graph, Gibbs was able to determine three states of equilibrium, i.e., "necessarily stable", "neutral", and "unstable", and whether or not changes would ensue.
Hence, in 1882, the German scientist Hermann von Helmholtz stated that affinity is the largest quantity of work which can be gained when the reaction is carried out in a reversible manner, e.g., electrical work in a reversible cell. The maximum work is thus regarded as the diminution of the free, or available, energy of the system (Gibbs free energy G at T = constant, P = constant or Helmholtz free energy F at T = constant, V = constant), whilst the heat given out is usually a measure of the diminution of the total energy of the system (Internal energy). Thus, G or F is the amount of energy “free” for work under the given conditions.
Up until this point, the general view had been such that: “all chemical reactions drive the system to a state of equilibrium in which the affinities of the reactions vanish”. Over the next 60 years, the term affinity came to be replaced with the term free energy. According to chemistry historian Henry Leicester, the influential 1923 textbook Thermodynamics and the Free Energy of Chemical Reactions by Gilbert N. Lewis and Merle Randall led to the replacement of the term “affinity” by the term “free energy” in much of the English-speaking world.
Graphical interpretation
Gibbs free energy was originally defined graphically. In 1873, American engineer Willard Gibbs published his first thermodynamics paper, “Graphical Methods in the Thermodynamics of Fluids”, in which Gibbs used the two coordinates of the entropy and volume to represent the state of the body. In his second follow-up paper, “A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces”, published later that year, Gibbs added in the third coordinate of the energy of the body, defined on three figures. In 1874, Scottish physicist James Clerk Maxwell used Gibbs' figures to make a 3D energy-entropy-volume thermodynamic surface of a fictitious water-like substance.[6] Thus, in order to understand the very difficult concept of Gibbs free energy one must be able to understand its interpretation as Gibbs defined originally by section AB on his figure 3 and as Maxwell sculpted that section on his 3D surface figure.
 American engineer Willard Gibbs' 1873 figures two and three (above left and middle) used by Scottish physicist James Clerk Maxwell in 1874 to create a three-dimensional entropy (x), volume (y), energy (z) thermodynamic surface diagram for a fictitious water-like substance, transposed the two figures of Gibbs (above right) onto the volume-entropy coordinates (transposed to bottom of cube) and energy-entropy coordinates (flipped upside down and transposed to back of cube), respectively, of a three-dimensional Cartesian coordinates; the region AB being the first-ever three-dimensional representation of Gibbs free energy, or what Gibbs called "available energy"; the region AC being its capacity for entropy, what Gibbs defined as "the amount by which the entropy of the body can be increased without changing the energy of the body or increasing its volume.
American engineer Willard Gibbs' 1873 figures two and three (above left and middle) used by Scottish physicist James Clerk Maxwell in 1874 to create a three-dimensional entropy (x), volume (y), energy (z) thermodynamic surface diagram for a fictitious water-like substance, transposed the two figures of Gibbs (above right) onto the volume-entropy coordinates (transposed to bottom of cube) and energy-entropy coordinates (flipped upside down and transposed to back of cube), respectively, of a three-dimensional Cartesian coordinates; the region AB being the first-ever three-dimensional representation of Gibbs free energy, or what Gibbs called "available energy"; the region AC being its capacity for entropy, what Gibbs defined as "the amount by which the entropy of the body can be increased without changing the energy of the body or increasing its volume.
Definitions
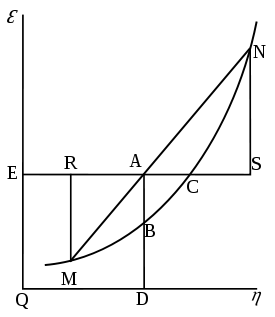 Willard Gibbs’ 1873 available energy (free energy) graph, which shows a plane perpendicular to the axis of v (volume) and passing through point A, which represents the initial state of the body. MN is the section of the surface of dissipated energy. Qε and Qη are sections of the planes η = 0 and ε = 0, and therefore parallel to the axes of ε (internal energy) and η (entropy), respectively. AD and AE are the energy and entropy of the body in its initial state, AB and AC its available energy (Gibbs free energy) and its capacity for entropy (the amount by which the entropy of the body can be increased without changing the energy of the body or increasing its volume) respectively.
Willard Gibbs’ 1873 available energy (free energy) graph, which shows a plane perpendicular to the axis of v (volume) and passing through point A, which represents the initial state of the body. MN is the section of the surface of dissipated energy. Qε and Qη are sections of the planes η = 0 and ε = 0, and therefore parallel to the axes of ε (internal energy) and η (entropy), respectively. AD and AE are the energy and entropy of the body in its initial state, AB and AC its available energy (Gibbs free energy) and its capacity for entropy (the amount by which the entropy of the body can be increased without changing the energy of the body or increasing its volume) respectively.The Gibbs free energy is defined as:
- G(p,T) = U + pV − TS
which is the same as:
- G(p,T) = H − TS
where:
- U is the internal energy (SI unit: joule)
- p is pressure (SI unit: pascal)
- V is volume (SI unit: m3)
- T is the temperature (SI unit: kelvin)
- S is the entropy (SI unit: joule per kelvin)
- H is the enthalpy (SI unit: joule)
The expression for the infinitesimal reversible change in the Gibbs free energy as a function of its 'natural variables' p and T, for an open system, subjected to the operation of external forces (for instance electrical or magnetical) Xi, which cause the external parameters of the system ai to change by an amount dai, can be derived as follows from the First Law for reversible processes:




where:
- μi is the chemical potential of the ith chemical component. (SI unit: joules per particle[7] or joules per mole[2])
- Ni is the number of particles (or number of moles) composing the ith chemical component.
This is one form of Gibbs fundamental equation.[8] In the infinitesimal expression, the term involving the chemical potential accounts for changes in Gibbs free energy resulting from an influx or outflux of particles. In other words, it holds for an open system. For a closed system, this term may be dropped.
Any number of extra terms may be added, depending on the particular system being considered. Aside from mechanical work, a system may, in addition, perform numerous other types of work. For example, in the infinitesimal expression, the contractile work energy associated with a thermodynamic system that is a contractile fiber that shortens by an amount −dl under a force f would result in a term fdl being added. If a quantity of charge −de is acquired by a system at an electrical potential Ψ, the electrical work associated with this is −Ψde, which would be included in the infinitesimal expression. Other work terms are added on per system requirements.[9]
Each quantity in the equations above can be divided by the amount of substance, measured in moles, to form molar Gibbs free energy. The Gibbs free energy is one of the most important thermodynamic functions for the characterization of a system. It is a factor in determining outcomes such as the voltage of an electrochemical cell, and the equilibrium constant for a reversible reaction. In isothermal, isobaric systems, Gibbs free energy can be thought of as a "dynamic" quantity, in that it is a representative measure of the competing effects of the enthalpic and entropic driving forces involved in a thermodynamic process.
The temperature dependence of the Gibbs energy for an ideal gas is given by the Gibbs-Helmholtz equation and its pressure dependence is given by:
if the volume is known rather than pressure then it becomes:
or more conveniently as its chemical potential:
In non-ideal systems, fugacity comes into play.
Derivation
The Gibbs free energy total differential natural variables may be derived via Legendre transforms of the internal energy.
 .
.
Because S, V, and Ni are extensive variables, Euler's homogeneous function theorem allows easy integration of dU:[10]
 .
.
The definition of G from above is
 .
.
Taking the total differential, we have
 .
.
Replacing dU with the result from the first law gives[10]

 .
.
The natural variables of G are then p, T, and {Ni}. Because some of the natural variables are intensive, dG may not be integrated using Euler integrals as is the case with internal energy. However, simply substituting the result for U into the definition of G gives a standard expression for G:[10]

 .
.
Free energy of reactions
To derive the Gibbs free energy equation for an isolated system, let Stot be the total entropy of the isolated system, that is, a system that cannot exchange heat or mass with its surroundings. According to the second law of thermodynamics:
and if ΔStot = 0 then the process is reversible. The heat transfer Q vanishes for an adiabatic system. Any adiabatic process that is also reversible is called an isentropic
 process.
process.Now consider systems, having internal entropy Sint. Such a system is thermally connected to its surroundings, which have entropy Sext. The entropy form of the second law applies only to the closed system formed by both the system and its surroundings. Therefore a process is possible if
 .
.
If Q is heat transferred to the system from the surroundings, so −Q is heat lost by the surroundings
- so that
 corresponds to entropy change of the surroundings.
corresponds to entropy change of the surroundings.
- We now have:

- Multiply both sides by T:

Q is heat transferred to the system; if the process is now assumed to be isobaric, then Qp = ΔH:
ΔH is the enthalpy change of reaction (for a chemical reaction at constant pressure). Then
for a possible process. Let the change ΔG in Gibbs free energy be defined as
 (eq.1)
(eq.1)
Notice that it is not defined in terms of any external state functions, such as ΔSext or ΔStot. Then the second law becomes, which also tells us about the spontaneity of the reaction:
 favoured reaction (Spontaneous)
favoured reaction (Spontaneous) Neither the forward nor the reverse reaction prevails (Equilibrium)
Neither the forward nor the reverse reaction prevails (Equilibrium) disfavoured reaction (Nonspontaneous)
disfavoured reaction (Nonspontaneous)
Gibbs free energy G itself is defined as
 (eq.2)
(eq.2)
but notice that to obtain equation (2) from equation (1) we must assume that T is constant. Thus, Gibbs free energy is most useful for thermochemical processes at constant temperature and pressure: both isothermal and isobaric. Such processes don't move on a P-V diagram, such as phase change of a pure substance, which takes place at the saturation pressure and temperature. Chemical reactions, however, do undergo changes in chemical potential, which is a state function. Thus, thermodynamic processes are not confined to the two dimensional P-V diagram. There is a third dimension for n, the quantity of gas. For the study of explosive chemicals, the processes are not necessarily isothermal and isobaric. For these studies, Helmholtz free energy is used.
If an isolated system (Q = 0) is at constant pressure (Q = ΔH), then
Therefore the Gibbs free energy of an isolated system is:
and if ΔG ≤ 0 then this implies that ΔS ≥ 0, back to where we started the derivation of ΔG
Useful identities
 for constant temperature
for constant temperature
 (see Chemical equilibrium).
(see Chemical equilibrium).

and rearranging gives
which relates the electrical potential of a reaction to the equilibrium coefficient for that reaction (Nernst equation).
where
ΔG = change in Gibbs free energy, ΔH = change in enthalpy, T = absolute temperature, ΔS = change in entropy, R = gas constant, ln = natural logarithm, ΔrG = change of reaction in Gibbs free energy, ΔrG° = standard change of reaction in Gibbs free energy, K = equilibrium constant, Qr = reaction quotient, n = number of electrons per mole product, F = Faraday constant (coulombs per mole), and E = electrode potential of the reaction. Moreover, we also have:
which relates the equilibrium constant with Gibbs free energy.
Standard energy change of formation
The standard Gibbs free energy of formation of a compound is the change of Gibbs free energy that accompanies the formation of 1 mole of that substance from its component elements, at their standard states (the most stable form of the element at 25 degrees Celsius and 100 kilopascals). Its symbol is ΔfG˚.
All elements in their standard states (oxygen gas, graphite, etc.) have 0 standard Gibbs free energy change of formation, as there is no change involved.
- ΔrG = ΔrG˚ + RT ln Qr; Qr is the reaction quotient.
At equilibrium, ΔrG = 0 and Qr = K so the equation becomes ΔrG˚ = −RT ln K; K is the equilibrium constant.
Table of selected substances[11]
Substance State ΔfG°(kJ/mol) ΔfG°(kcal/mol) NH3 g -12.67 −3.976 H2O l -237.13 −56.69 H2O g -174.13 −54.64 CO2 g -300.39 −94.26 CO g -104.56 −32.81 CH4 g -38.69 −12.14 C2H6 g -25.05 −7.86 C3H8 g -17.89 −5.614 C8H18 g 13.19 4.14 C10H22 g 26.23 8.23 See also
References
- ^ Greiner, Walter; Neise, Ludwig; Stöcker, Horst (1995). Thermodynamics and statistical mechanics. Springer-Verlag. p. 101.
- ^ a b c Perrot, Pierre (1998). A to Z of Thermodynamics. Oxford University Press. ISBN 0-19-856552-6.
- ^ J.W. Gibbs, “A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces,” Transactions of the Connecticut Academy of Arts and Sciences 2, Dec. 1873, pp. 382-404 (quotation on p. 400).
- ^ Reiss, Howard (1965). Methods of Thermodynamics. Dover Publications. ISBN 0-486-69445-3.
- ^ International Union of Pure and Applied Chemistry Commission on Atomspheric Chemistry, J. G. (1990). "Glossary of Atmospheric Chemistry Terms (Recommendations 1990)". Pure Appl. Chem. 62 (11): 2167–2219. doi:10.1351/pac199062112167. http://www.iupac.org/publications/pac/1990/pdf/6211x2167.pdf. Retrieved 2006-12-28. International Union of Pure and Applied Chemistry Commission on Physicochemical Symbols Terminology and Units (1993). Quantities, Units and Symbols in Physical Chemistry (2nd Edition). Oxford: Blackwell Scientific Publications. pp. 48. ISBN 0-632-03583-8. http://www.iupac.org/publications/books/gbook/green_book_2ed.pdf. Retrieved 2006-12-28. International Union of Pure and Applied Chemistry Commission on Quantities and Units in Clinical Chemistry, H. P.; International Federation of Clinical Chemistry and Laboratory Medicine Committee on Quantities and Units (1996). "Glossary of Terms in Quantities and Units in Clinical Chemistry (IUPAC-IFCC Recommendations 1996)". Pure Appl. Chem. 68 (4): 957–1000. doi:10.1351/pac199668040957. http://www.iupac.org/publications/pac/1996/pdf/6804x0957.pdf. Retrieved 2006-12-28.
- ^ James Clerk Maxwell, Elizabeth Garber, Stephen G. Brush, and C. W. Francis Everitt (1995), Maxwell on heat and statistical mechanics: on "avoiding all personal enquiries" of molecules, Lehigh University Press, ISBN 093422334, p. 248.
- ^ Chemical Potential - IUPAC Gold Book
- ^ Müller, Ingo (2007). A History of Thermodynamics - the Doctrine of Energy and Entropy. Springer. ISBN 978-3-540-46226-2.
- ^ Katchalsky, A.; Curran, Peter, F. (1965). Nonequilibrium Thermodynamics in Biophysics. Harvard University Press. CCN 65-22045.
- ^ a b c Salzman, William R. (2001-08-21). "Open Systems". Chemical Thermodynamics. University of Arizona. Archived from the original on 2007-07-07. http://web.archive.org/web/20070707224025/http://www.chem.arizona.edu/~salzmanr/480a/480ants/opensys/opensys.html. Retrieved 2007-10-11.
- ^ Handbook of chemistry and physics, 1960, p.1882-1915, p.1919-1921, 42nd ed., Harrison
External links
- IUPAC definition (Gibbs energy)
- Gibbs energy - Florida State University
- Gibbs Free Energy - Eric Weissteins World of Physics
- Entropy and Gibbs Free Energy - www.2ndlaw.oxy.edu
- Gibbs Free Energy - Georgia State University
- Gibbs Free Energy Java Applet - University of California, Berkeley
- Gibbs Free Energy - Illinois State University
- Using Gibbs Free Energy for prediction of chemical driven material ageing
Categories:- Fundamental physics concepts
- State functions
- Thermodynamic free energy



















Wikimedia Foundation. 2010.