- CCR5 receptor antagonists
-
CCR5 receptor antagonists are a class of small molecules that antagonize the CCR5 receptor. The C-C motif chemokine receptor CCR5 is involved in the HIV entry process. Hence antagonists of this receptor have potential therapeutic applications in the treatment of HIV infections.
The life cycle of the Human Immunodeficiency Virus (HIV) presents potential targets for drug therapy, one of them being the viral entry pathway. The C-C motif chemokine receptors CCR5 and CXCR4 are the main chemokine receptors involved in the HIV entry process. These receptors belong to the seven transmembrane G-protein-coupled receptor (GPCR) family and are predominantly expressed on human T-cells, dendritic cells and macrophages.[1] They play an important role as co-receptors that HIV type 1 (HIV-1) uses to attach to cells before viral fusion and entry into host cells.[1] HIV isolates can be divided into R5 and X4 strains. R5 strain is when the virus uses the co-receptor CCR5 and X4 strain is when it uses CXCR4.[2] The location of CCR5 receptors at the cell surface, both large and small molecules have the potential to interfere with the CCR5-viral interaction and inhibit viral entry into human cells.[3]
Contents
History
Since the discovery of HIV in the 1980s, remarkable progress has been made in the development of novel antiviral drugs.[2] The trigger for the discovery of the CCR5 antagonists was the observation that a small percentage of high-risk populations showed either resistance or delayed development of the disease. This population was found to have a mutation (CCR5-Δ32) in the gene that codes for the CCR5 receptor which results in almost complete resistance against HIV-1 infection and scientists then discovered the key role of the cell surface receptors CCR5 and CXCR4 in successful viral fusion and infection.[4] In 1996, it was demonstrated that CCR5 serves as a co-receptor for the most commonly transmitted HIV-1 strains, R5. This type of virus is predominant during the early stages of infection and remains the dominant form in over 50% of late stage HIV-1 infected patients,[5][6] however R5 strains can eventually evolve into X4 as the disease progresses.[2] This information led to the development of a new class of HIV drugs called CCR5 antagonists.[7]
Mechanism of action
 Figure 1 HIV entry into CD4+ cell via CCR5 co-receptor.
Figure 1 HIV entry into CD4+ cell via CCR5 co-receptor.
HIV enters host cells in the blood by attaching itself to receptors on the surface of the CD4+ cell.[8] Viral entry to the CD4+ cell begins with attachment of the R5 HIV-1 glycoprotein 120 (gp120) to the CD4+ T-cell receptor, which produces a conformational change in gp120 and allows it to bind to CCR5, thereby triggering glycoprotein 41 (gp41) mediated fusion of the viral envelope with the cell membrane and the nucleocapsid enters the host cell (Figure 1).[8][9] CCR5 co-receptor antagonists prevent HIV-1 from entering and infecting immune cells by blocking CCR5 cell-surface receptor.[10] Small molecule antagonists of CCR5 bind to a hydrophobic pocket formed by the transmembrane helices of the CCR5 receptor.[11] They are thought to interact with the receptor in an allosteric manner locking the receptor in a conformation that prohibits its co-receptor function.[12]
Drug development
As mentioned, the CCR5 receptor is a G-protein coupled receptor (GPCR) and before the discovery of CCR5’s role in HIV infection many pharmaceutical companies had already built a substantial collection of compounds that target GPCRs. Some of these compounds provided an excellent starting point for CCR5 antagonists but then needed further optimization to gain better potency, pharmacokinetic properties and better CCR5 selectivity. One of the biggest problems was the affinity for the hERG ion channel, as inhibition of hERG can lead to QT interval prolongation. Abnormality in the interval can increase the risk of developing ventricular arrhythmias.[3][13] Many CCR5 antagonists have been introduced by pharmaceutical companies but few of them have actually reached human efficacy studies, for example AstraZeneca, Novartis, Merck and Takeda have used these GPRC compound collections to develop a potent CCR5 antagonist but none of them have reached clinical trials.[14][15][16][17] Three pharmaceutical companies were racing for the first approved small molecule CCR5 antagonist; GlaxoSmithKline(GSK), with their compound aplaviroc, Schering-Plough with vicriviroc and Pfizer with maraviroc. All the compounds reached clinical trials in humans but only maraviroc has been approved by the U.S. Food and Drug Administration (FDA).[3] In the following section the development of these three compounds will be discussed.
Aplaviroc
 Figure 2 Molecular structure of aplaviroc and its lead compound
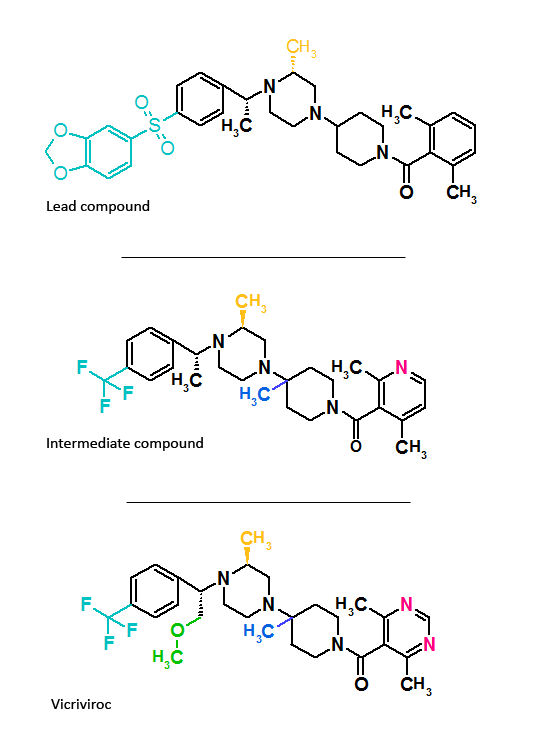
Figure 2 Molecular structure of aplaviroc and its lead compound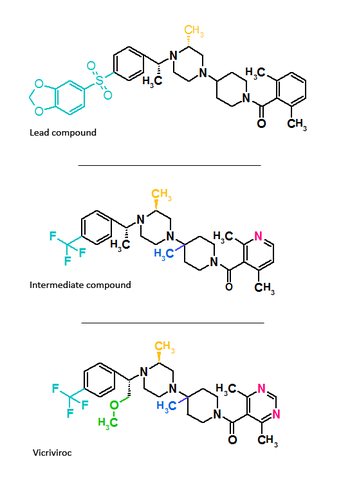 Figure 3 Molecular structure of vicriviroc and its lead compounds
Figure 3 Molecular structure of vicriviroc and its lead compoundsAplaviroc is originated from a class of spirodiketopiperazine derivatives. Figure 2 shows the molecular structure of the lead compound and the final compound aplaviroc. The lead compound showed good potency in blocking CCR5 in a number of R5 HIV strains and against multi-drug resistant strains.[3] The problem with this compound was not its CCR5 selectivity but the oral bioavailability.[3][18] This led to further development of the molecule and the result was a compound named aplaviroc. Unfortunately despite the promising preclinical and early clinical results some severe liver toxicity was observed in the treatment of naive and treatment-experienced patients that led to the discontinuation in further development of aplaviroc.[3]
Vicriviroc
Schering-Plough identified an active compound during screening.[3] Figure 3 shows the molecular structure of the lead compound, intermediate compound and the final compound vicriviroc. The lead compound contained a piperazine scaffold and was a potent muscarinic acetylcholine receptor (M2) antagonist with modest CCR5 activity. The changes that were made on the left hand side of the lead compound and the addition of a methyl group on the piperazine group ((S)-methylpiperazine) resulted in the intermediate compound that had good affinity for CCR5 receptors but very little affinity for muscarinic activity, however, the compound did show affinity for the hERG ion channel.[19][20] Further reconstruction led to the development of the final compound vicriviroc, when Schering discovered that the pyridyl N-oxide on the intermediate could be replaced by 4,6-dimethylpyrimidine carboxamide. Vicriviroc had an excellent selectivity for CCR5 receptors over muscarinic and hERG affinity was greatly reduced.[21][22] Phase I clinical trial of vicriviroc gave promising results, so a phase II study in the treatment of naive patients was initiated. The phase II study was discontinued since there was a viral breakthrough in the vicriviroc group compared to the control group. These results suggested that vicriviroc was not effective in the treatment of treatment-naive patients compared to current treatments. Another phase II clinical study was performed in treatment-experienced patients. The results were that vicriviroc did have strong antiviral activity but five instances of cancer among the participants were reported, however, the study was continued since there was lack of causal association of the malignancies and vicriviroc.[3] In late 2009 vicriviroc was assigned in phase III studies in treatment-experienced patients, and in phase II studies in treatment for naive patients.[23]
Maraviroc
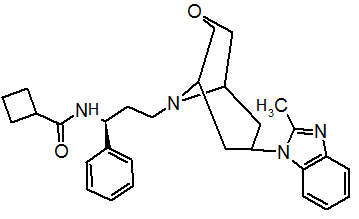
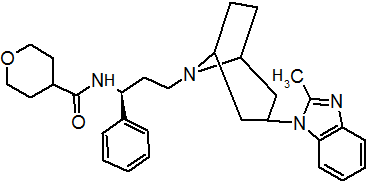
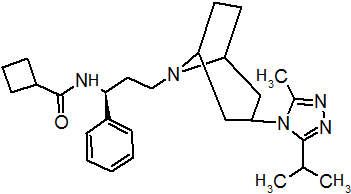
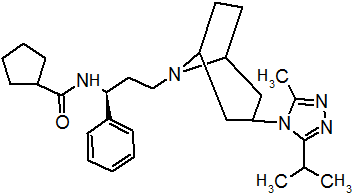
Pfizer turned to high-throughput screening in their search for a good starting point for a small molecule CCR5 antagonist. Their screening resulted in a compound that presented weak affinity and no antiviral activity but represented a good starting point for further optimization.[3] Compounds 1-9 in table 1 show the development of maraviroc in few steps. The chemical structure of the starting molecule is presented as compound 1. Their first focus was to minimize CYP2D6 activity of the molecule and to reduce its lipophilicity. They replaced the imidazopyridine with benzimidazole and the benzhydril group was swapped out for a benzamide. The outcome was compound 2.[3] That compound showed good binding potency and the start of an antiviral activity. Further SAR (structure-activity relationship) optimization of the amide region and identifying the enantiomeric preference led to the cyclobutyl amide structure in compound 3. However, the problem with the CYP2D6 activity of the compound was still unacceptable so they had to perform further SAR optimization that determined that the [3.2.1]-azabicycloamine (topane) could replace the aminopiperidine moiety. This change in the chemical structure led to compound 4. Compound 4 had no CYP2D6 activity while preserving excellent binding affinity and antiviral activity.[3][24] Although compound 4 showed promising results it demonstrated 99% inhibition on the hERG ion channel. That inhibition was unacceptable since it can lead to QTc interval prolongation. The research team then did a few modifications to see which part of the molecule played a role in the hERG affinity. Compound 5 shows an analogue that they synthesized which contained an oxygen bridgehead in the tropane ring; however, that reconstruction did not have an effect on the hERG affinity.[25] They then focused on the polar surface area in the molecule to dial out the hERG affinity. These efforts resulted in compound 6. That compound preserved desired antiviral activity and was selective against the hERG inhibition but the problem was its bioavailability. Reduction in the lipophilicity, by replacing the benzimidazol group with a substituted triazole group gave compound 7. Compound 7 had shown a significant reduction in lipophilicity and maintained the antiviral activity but again, with the introduction of a cyclobutyl group the compound showed hERG inhibition. Changing the ring size in compound 7 from a cyclobutyl unit to a cyclopentyl unit in compound 8 led to a significant increase in antiviral activity and loss of hERG affinity. Further development led to discovery of a 4,4’-difluorocyclohexylamide also known as maraviroc. Maraviroc preserved excellent antiviral activity, whilst demonstrating no significant hERG binding affinity. The lack of hERG binding affinity was predicted to be because of the large size of the cyclohexyl group and the high polarity of the fluoro substituents.[3][24][25] In August 2007 the FDA approved the first CCR5 antagonist, maraviroc discovered and developed by Pfizer.[4][7]
Table 1 Represents the molecular structures in the development of maraviroc 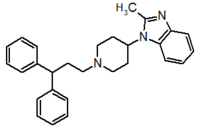
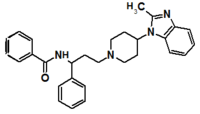
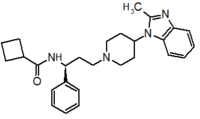
Compound 1 Compound 2 Compound 3 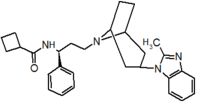
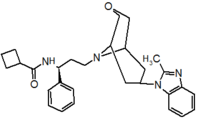
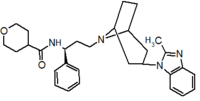
Compound 4 Compound 5 Compound 6 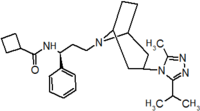
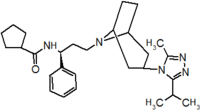
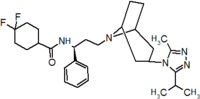
Compound 7 Compound 8 Compound 9 (Maraviroc) Pharmacophore
 Figure 4 Predictive pharmacophore model for piperidine- and piperazine-based CCR5 antagonists
Figure 4 Predictive pharmacophore model for piperidine- and piperazine-based CCR5 antagonistsThe predictive pharmacophore model was developed for a large series of piperidine- and piperazine-based CCR5 antagonists by Schering-Plough Research Institute. Their hypothesis consisted of mostly five features, two hydrogen bond acceptors, marked C and D in figure 4 and three hydrophobic groups, A, B and E in figure 4. Part B usually has a basic nitrogen group. The model was validated using diverse set of six CCR5 antagonists from five different pharmaceutical companies. The best model correctly predicted these compounds as being highly active. It is possible to use the model as a tool in virtual screening for new small molecular CCR5 antagonists and also to predict biological activities of compounds prior to undertaking their costly synthesis.[26]
Binding
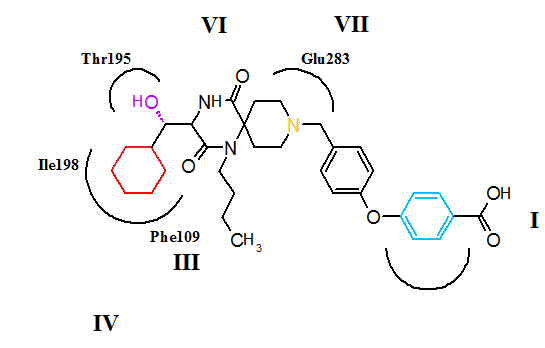
CCR5 is a member of G-protein-coupled, seven transmembrane segment receptors. The structure of the receptor comprises seven-helix bundle in the transmembrane region, these regions are labeled I-VII in figure 5 and 6. The CCR5 antagonists are predicted to bind to a putative binding pocket which is buried inside the transmembrane domain, enclosed by the seven transmembrane helices. The binding pocket is very hydrophobic with multiple aromatic residues lining the pocket. The key residues are tryptophan 86 and 248 (Trp86, Trp248), tyrosine 108 and 251 (Tyr108, Tyr251), phenylalanine 109 (phe109), threonine 195 (Thr195), isoleucine 198 (Ile198), glutamic acid 283 (Glu283). CCR5 antagonists are very different in shape and electrostatic potential although they all share the same binding pocket. The interesting thing about the binding of these molecules is that they exhibit significantly different binding modes, although they all establish an extensive interaction network with CCR5.[27][28][29]
Aplaviroc
 Figure 5 Putative binding of aplaviroc to the CCR5 receptor
Figure 5 Putative binding of aplaviroc to the CCR5 receptor Figure 6 Putative binding of maraviroc to the CCR5 receptor
Figure 6 Putative binding of maraviroc to the CCR5 receptorThe putative binding mode for aplaviroc is shown in figure 5. The key saltbridge interaction between aplaviroc and Glu283 is predicted to be quite weak compared to other CCR5 antagonists. The hydroxyl group on aplaviroc forms a strong hydrogen bond to the polar residue Thr195. This H-bond interaction is the strongest with aplaviroc compared to other CCR5 antagonists. The cyclohexyl group in the aplaviroc structure is predicted to interact with the receptor in a hydrophobic pocket formed by Ile198, Thr15 and Phe109 and is thought to show quite strong hydrophobic interactions. The researchers predict that the t-butyl group of aplaviroc is buried within the helical bundle through strong hydrophobic interaction with multiple aromatic residues of the CCR5 receptor.[28]
Maraviroc
The putative binding mode for maraviroc is shown in figure 6. The strongest interaction is estimated to be between maraviroc and glutamic acid (Glu283) through a strong salt-bridge interaction. The interaction between tryptophan (Trp86) and maraviroc involves T-shaped π-π stacking while the interaction with phenylalanine (Phe109) is predicted to be hydrophopic. Tyrosine (Tyr108) is thought to interact with the phenyl group on maraviroc through a parallel displaced interaction. The interaction between maraviroc and isoleucine (Ile198) is predicted to be mostly hydrophobic in nature and the interaction between maraviroc and tyrosine (Tyr251) is very limited.[28]
Aplaviroc
Aplaviroc has a unique feature of preserving two of the natural chemokine protein ligands binding to CCR5 and subsequent activation, whereas maraviroc and the other antagonists almost fully block chemokine-CCR5 interactions. This kind of interference is so far considered to be safe, and individuals that naturally lack CCR5 do not show any obvious health problems. However, to limit the toxicity and side effects of CCR5 antagonists it would be ideal to be able to preserve the chemokine receptor function. Consequently, it should be of interest to design inhibitors that specifically disrupt CCR5-gp120 binding but do not affect the CCR5 chemokine activation.[29]
Other CCR5 antagonists
 Figure 7 Molecular structure of compound A
Figure 7 Molecular structure of compound ADevelopment of new CCR5 antagonists continues, both for their antiviral effects and also for potential utility in a variety of autoimmune indications. Researchers at Ono have discovered a novel series of potent CCR5 small molecule antagonists. Lead optimization was pursued by balancing opposing trends of metabolic stability and potency. Combination of the spiropiperidine template with pharmacophore elements from both aplaviroc, and Schering’s CCR5 antagonist program, led to the initial lead compound in this series. Further development of that lead compound led to the discovery of compound A in figure 7 - A compound that possesses a good selectivity and pharmacokinetic properties.[30]
The CCR5 antagonist INCB009471 has nanomolar activity against HIV-1 in vitro. This compound demonstrated potent and prolonged antiviral activity against R5-tropic HIV-1 when given 200 mg once daily dose for 14 days. These findings supported further clinical development of INCB009471 and they have since progressed to phase IIb clinical trials. As of 2009 the study of this compound is inactive and no further studies are planned at this time.[31]
See also
- Cenicriviroc
- CD4
- CCL5
- Subtypes of HIV
- HIV tropism
- Discovery and development of non-nucleoside reverse transcriptase inhibitors
- Discovery and development of nucleoside and nucleotide reverse-transcriptase inhibitors
References
- ^ a b Lederman MM, Penn-Nicholson A, Cho M, Mosier D (August 2006). "Biology of CCR5 and its role in HIV infection and treatment". JAMA 296 (7): 815–26. doi:10.1001/jama.296.7.815. PMID 16905787.
- ^ a b c De Clercq E (December 2007). "The design of drugs for HIV and HCV". Nature Reviews. Drug Discovery 6 (12): 1001–18. doi:10.1038/nrd2424. PMID 18049474.
- ^ a b c d e f g h i j k l Pulley, Shon (2007). [10.1007/978-3-7643-7437-2_11 "CCR5 antagonists: from discovery to clinical efficacy"]. In Neote, Kuldeep; Letts, Gordon L.; Moser, Bernhard. Chemokine Biology — Basic Research and Clinical Application. 2. Birkhauser Basel. pp. 145–163. ISBN 978-3-7643-7195-2. 10.1007/978-3-7643-7437-2_11.
- ^ a b Lalezari, J.; Goodrich, J.; DeJesus, E.; Lampiris, H.; Gulick, R.; Saag, M.; Ridgway, C.; McHale, M. et al.. "Efficacy and Safety of Maraviroc plus Optimized Background Therapy In Viremic, ART-Experienced Patients Infected With CCR5-Tropic HIV‑1, 24-week results of a Phase 2b/3 study in the U. S. & Canada. Abstract 104bLB". 14th Conference on Retroviruses and Opportunistic Infections. http://www.retroconference.org/2007/abstracts/30635.htm.
- ^ Samson M, Libert F, Doranz BJ, et al. (August 1996). "Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene". Nature 382 (6593): 722–5. doi:10.1038/382722a0. PMID 8751444.
- ^ Dragic T, Litwin V, Allaway GP, et al. (June 1996). "HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5". Nature 381 (6584): 667–73. doi:10.1038/381667a0. PMID 8649512.
- ^ a b Flexner C (December 2007). "HIV drug development: the next 25 years". Nature Reviews. Drug Discovery 6 (12): 959–66. doi:10.1038/nrd2336. PMID 17932493.
- ^ a b Ray N, Doms RW (2006). "HIV-1 coreceptors and their inhibitors". Current Topics in Microbiology and Immunology. Current Topics in Microbiology and Immunology 303: 97–120. doi:10.1007/978-3-540-33397-5_5. ISBN 978-3-540-29207-4. PMID 16570858.
- ^ Westby M, van der Ryst E (2005). "CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection". Antiviral Chemistry & Chemotherapy 16 (6): 339–54. PMID 16329283.
- ^ Briz V, Poveda E, Soriano V (April 2006). "HIV entry inhibitors: mechanisms of action and resistance pathways". The Journal of Antimicrobial Chemotherapy 57 (4): 619–27. doi:10.1093/jac/dkl027. PMID 16464888.
- ^ Murga JD, Franti M, Pevear DC, Maddon PJ, Olson WC (October 2006). "Potent Antiviral Synergy between Monoclonal Antibody and Small-Molecule CCR5 Inhibitors of Human Immunodeficiency Virus Type 1". Antimicrobial Agents and Chemotherapy 50 (10): 3289–96. doi:10.1128/AAC.00699-06. PMC 1610098. PMID 17005807. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1610098.
- ^ Watson C, Jenkinson S, Kazmierski W, Kenakin T (April 2005). "The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor". Molecular Pharmacology 67 (4): 1268–82. doi:10.1124/mol.104.008565. PMID 15644495.
- ^ Fermini B, Fossa AA (June 2003). "The impact of drug-induced QT interval prolongation on drug discovery and development". Nature Reviews. Drug Discovery 2 (6): 439–47. doi:10.1038/nrd1108. PMID 12776219.
- ^ Cumming JG, Cooper AE, Grime K, et al. (November 2005). "Modulators of the human CCR5 receptor. Part 2: SAR of substituted 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides". Bioorganic & Medicinal Chemistry Letters 15 (22): 5012–5. doi:10.1016/j.bmcl.2005.08.014. PMID 16154744.
- ^ Dorn CP, Finke PE, Oates B, et al. (January 2001). "Antagonists of the human CCR5 receptor as anti-HIV-1 agents. part 1: discovery and initial structure-activity relationships for 1 -amino-2-phenyl-4-(piperidin-1-yl)butanes". Bioorganic & Medicinal Chemistry Letters 11 (2): 259–64. doi:10.1016/S0960-894X(00)00637-5. PMID 11206473.
- ^ Thoma G, Nuninger F, Schaefer M, et al. (April 2004). "Orally bioavailable competitive CCR5 antagonists". Journal of Medicinal Chemistry 47 (8): 1939–55. doi:10.1021/jm031046g. PMID 15055994.
- ^ Tremblay CL, Giguel F, Guan Y, Chou TC, Takashima K, Hirsch MS (August 2005). "TAK-220, a Novel Small-Molecule CCR5 Antagonist, Has Favorable Anti-Human Immunodeficiency Virus Interactions with Other Antiretrovirals In Vitro". Antimicrobial Agents and Chemotherapy 49 (8): 3483–5. doi:10.1128/AAC.49.8.3483-3485.2005. PMC 1196290. PMID 16048964. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1196290.
- ^ Maeda K, Nakata H, Koh Y, et al. (August 2004). "Spirodiketopiperazine-Based CCR5 Inhibitor Which Preserves CC-Chemokine/CCR5 Interactions and Exerts Potent Activity against R5 Human Immunodeficiency Virus Type 1 In Vitro". Journal of Virology 78 (16): 8654–62. doi:10.1128/JVI.78.16.8654-8662.2004. PMC 479103. PMID 15280474. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=479103.
- ^ Tagat JR, McCombie SW, Steensma RW, et al. (August 2001). "Piperazine-based CCR5 antagonists as HIV-1 inhibitors. I: 2(S)-methyl piperazine as a key pharmacophore element". Bioorganic & Medicinal Chemistry Letters 11 (16): 2143–6. doi:10.1016/S0960-894X(01)00381-X. PMID 11514156.
- ^ Tagat JR, Steensma RW, McCombie SW, et al. (October 2001). "Piperazine-based CCR5 antagonists as HIV-1 inhibitors. II. Discovery of 1-[(2,4-dimethyl-3-pyridinyl)carbonyl]-4- methyl-4-[3(S)-methyl-4-[1(S)-[4-(trifluoromethyl)phenyl]ethyl]-1-piperazinyl]- piperidine N1-oxide (Sch-350634), an orally bioavailable, potent CCR5 antagonist". Journal of Medicinal Chemistry 44 (21): 3343–6. doi:10.1021/jm0155401. PMID 11585438.
- ^ McCombie SW, Tagat JR, Vice SF, et al. (February 2003). "Piperazine-based CCR5 antagonists as HIV-1 inhibitors. III: synthesis, antiviral and pharmacokinetic profiles of symmetrical heteroaryl carboxamides". Bioorganic & Medicinal Chemistry Letters 13 (3): 567–71. doi:10.1016/S0960-894X(02)00918-6. PMID 12565973.
- ^ Tagat JR, McCombie SW, Nazareno D, et al. (May 2004). "Piperazine-based CCR5 antagonists as HIV-1 inhibitors. IV. Discovery of 1-[(4,6-dimethyl-5-pyrimidinyl)carbonyl]- 4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-methyl-1-piperazinyl]- 4-methylpiperidine (Sch-417690/Sch-D), a potent, highly selective, and orally bioavailable CCR5 antagonist". Journal of Medicinal Chemistry 47 (10): 2405–8. doi:10.1021/jm0304515. PMID 15115380.
- ^ "Schering-Plough Reports Long-Term Vicriviroc Data From Phase II Open-Label Extension Study in Treatment-Experienced HIV-Infected Patients" (Press release). Schering-Plough. 14 September 2009. http://www.natap.org/2009/ICCAC/ICCAC_20.htm. Retrieved 8 November 2009.
- ^ a b Wood A, Armour D (2005). "The discovery of the CCR5 receptor antagonist, UK-427,857, a new agent for the treatment of HIV infection and AIDS". Progress in Medicinal Chemistry. Progress in Medicinal Chemistry 43: 239–71. doi:10.1016/S0079-6468(05)43007-6. ISBN 9780444515728. PMID 15850827.
- ^ a b Price DA, Armour D, de Groot M, et al. (September 2006). "Overcoming HERG affinity in the discovery of the CCR5 antagonist maraviroc". Bioorganic & Medicinal Chemistry Letters 16 (17): 4633–7. doi:10.1016/j.bmcl.2006.06.012. PMID 16782336.
- ^ Debnath AK (October 2003). "Generation of predictive pharmacophore models for CCR5 antagonists: study with piperidine- and piperazine-based compounds as a new class of HIV-1 entry inhibitors". Journal of Medicinal Chemistry 46 (21): 4501–15. doi:10.1021/jm030265z. PMID 14521412.
- ^ Maeda K, Das D, Ogata-Aoki H, et al. (May 2006). "Structural and molecular interactions of CCR5 inhibitors with CCR5". The Journal of Biological Chemistry 281 (18): 12688–98. doi:10.1074/jbc.M512688200. PMID 16476734.
- ^ a b c Kondru R, Zhang J, Ji C, et al. (March 2008). "Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists". Molecular Pharmacology 73 (3): 789–800. doi:10.1124/mol.107.042101. PMID 18096812.
- ^ a b Wang T, Duan Y (June 2008). "Binding Modes of CCR5-targetting HIV Entry Inhibitors: Partial and Full Antagonists". Journal of Molecular Graphics & Modelling 26 (8): 1287–95. doi:10.1016/j.jmgm.2007.12.003. PMC 2701198. PMID 18249144. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2701198.
- ^ Rotstein DM, Gabriel SD, Makra F, et al. (September 2009). "Spiropiperidine CCR5 antagonists". Bioorganic & Medicinal Chemistry Letters 19 (18): 5401–6. doi:10.1016/j.bmcl.2009.07.122. PMID 19674898.
- ^ Kuritzkes DR (March 2009). "HIV-1 Entry Inhbitors: An Overview". Current Opinion in HIV and AIDS 4 (2): 82–7. doi:10.1097/COH.0b013e328322402e. PMC 2753507. PMID 19339945. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2753507.
Categories:- Drug discovery
- HIV/AIDS








Wikimedia Foundation. 2010.