- Discovery and development of renin inhibitors
-
Renin inhibitors are antihypertensive drugs that inhibit the first and rate-limiting step of the renin-angiotensin-aldosterone system (RAAS). Since the 1970s scientists have been trying to develop potent inhibitors with acceptable oral bioavailability.[1][2] The process was difficult and took about three decades. The first and second generations faced problems like poor bioavailability and lack of potency. Finally the third generation was discovered. These compounds were non-peptidic renin inhibitors, had acceptable oral bioavailability and were potent enough for clinical use. The first drug in this class was aliskiren which received a marketing approval in 2007.[1]
Contents
History
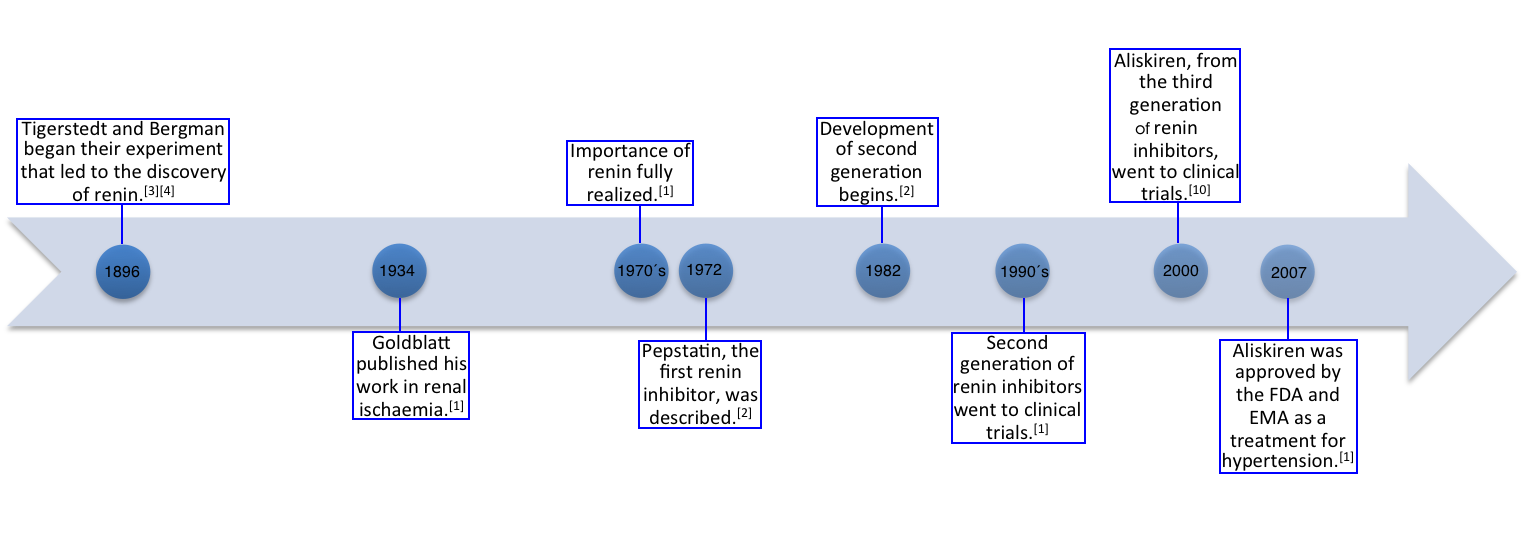
In 1896 the Finnish physiologist Robert Tigerstedt and the Swedish physician Per Bergman did an experiment on kidneys and the circulation in rabbits. They observed that blood pressure rose in the rabbits when extracts of the kidneys were injected into the jugular vein.[3][4] They also discovered that this substance that was responsible for higher blood pressure was produced in the renal cortex and they named this substance renin.[4] Although this experiment laid the foundation for future investigations into the RAAS pathway it had little impact on the scientific community at that time.[3][5] It was not until in 1934 when Goldblatt published his work in renal ischaemia that renin came into focus again. The importance of renin in the pathogenesis of cardiovascular disease was however not fully understood until in the 1970s and it was not until 20 years later that the first renin inhibitors went to clinical trials.[1]
Pepstatin, which was described in 1972, was the first synthetic renin inhibitor but poor pharmacokinetic properties prevented it from entering in vivo investigations.[2][6] The first generation of renin inhibitors, such as H-142, were peptide analogues of angiotensinogen.[7] However these inhibitors had also limited drug-like properties.[1][6] It was not until 1982 that hopes of breakthrough appeared when development of the second generation renin inhibitors begins.[1] This generation consisted of peptide-like compounds such as remikiren, enalkiren and zanikiren.[5] They had more drug-like rather than substrate-like properties and in 1990 they went to clinical trials. However the second generation had its limitations and never completed clinical trials.[1]
Aliskiren is a third generation renin inhibitor, which are non-peptide-like compounds, and is the only renin inhibitor that went into phase III clinical trials.[1][8] The first clinical trial of aliskiren was performed in 2000 in healthy volunteers.[9] In 2007 aliskiren was approved by the US Food and Drug Administration and the European Medicines Agency as a treatment for hypertension.[1]
 Timeline: Discovery and development of renin inhibitors
Timeline: Discovery and development of renin inhibitors
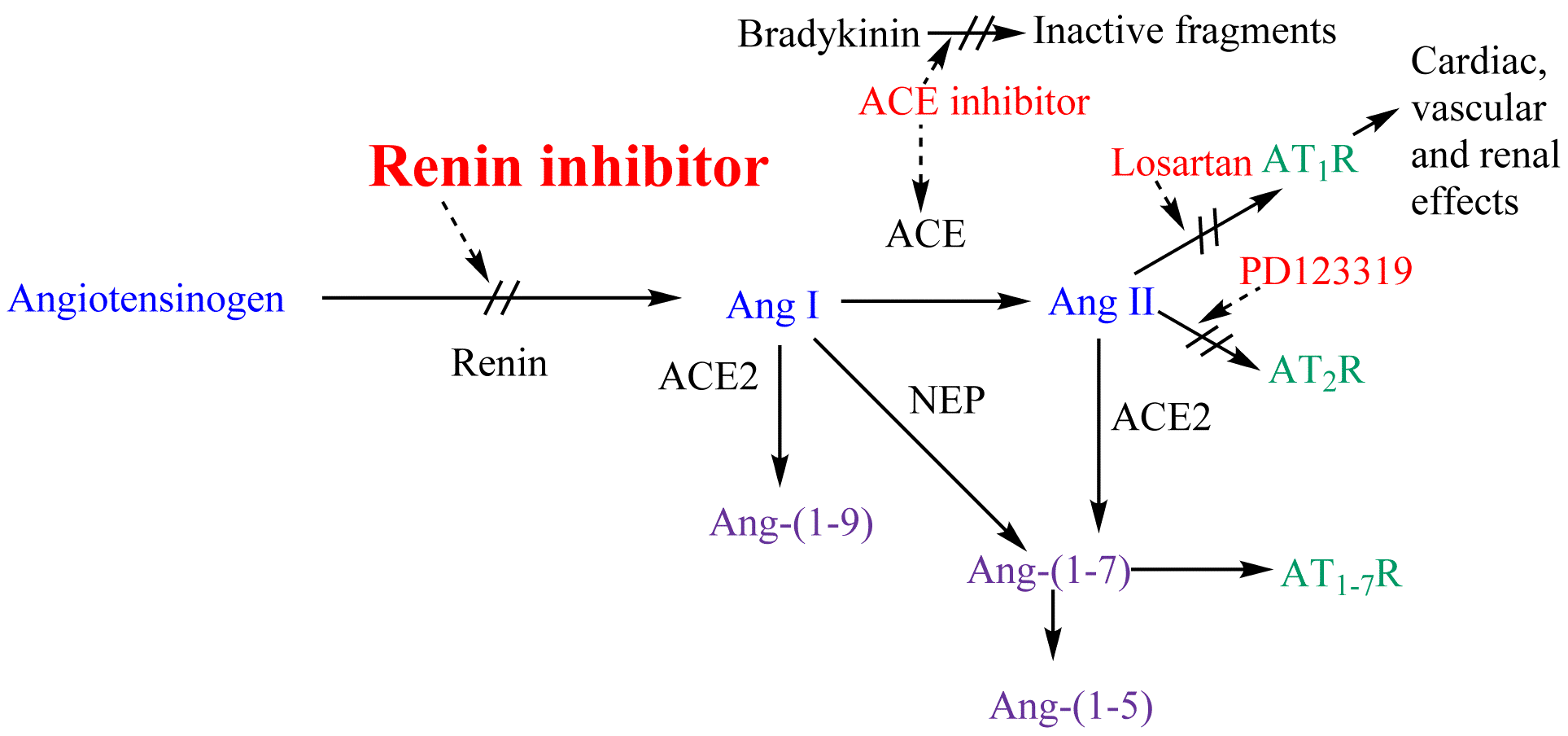
The renin-angiotensin-aldosterone system
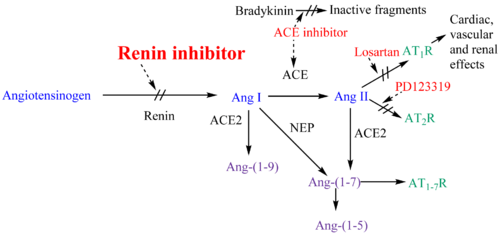 Renin-angiotensin-aldosterone system and potential steps of blockage
Renin-angiotensin-aldosterone system and potential steps of blockageThe renin-angiotensin-aldosterone system (RAAS) plays a key role in the pathology of hypertension. Under normal conditions, stimulation of the RAAS occurs in response to threats that compromise blood pressure stability, such as hypotension, blood loss and excessive loss of sodium and water. Blood pressure is a result of total peripheral resistance and cardiac output. Research has shown that renin is in high concentration when vasoconstriction is the main reason for high blood pressure. On the other hand, renin is in low concentration when cardiac output is the main reason.[10]
The highly selective aspartic protease renin is secreted from the juxtaglomerular apparatus as well as locally in other tissues, such as in the brain, heart and blood vessels.[11][12] Renin is a circulating enzyme that acts on a circulating peptide, angiotensinogen.[10] Renin cleaves the peptide at the Leu10–Val11 bond and this reaction is the rate-determining step of the RAAS.[13] This leads to the product angiotensin I (Ang I) which is a decapeptide. Ang I is broken down by the angiotensin-converting enzyme (ACE) to the active octapeptide angiotensin II (Ang II) which is the principal effector of the RAAS.[10] Ang II is a powerful vasoconstrictor and leads to the release of catecholamines from the adrenal medulla and prejunctional nerve endings.[14] Ang II also leads to the secretion of aldosterone which is the main salt-retaining hormone. This stimulates the kidneys to retain salt.[12] Ang II also provides a negative feedback to the system by its inhibiton on renin release.[14] Ang II interacts with at least two classes of Ang II receptors, AT1 and AT2.[12] This mechanism, which runs from renin through Ang II and to aldosterone as well as the negative feedback that Ang II has on renin secretion, is known as RAAS.[14] The short term effects are increased blood pressure and long term effects can be end organ damage.[10]
Mechanism of action
Renin inhibitors bind to the active site of renin and inhibit the binding of renin to angiotensinogen which is the rate determining step of the RAAS cascade.[10] By inhibiting the RAAS at the beginning renin inhibitors prevent the formation of Ang I and Ang II. It is also thought that renin inhibitors prevent Ang-(1-7), Ang-(1-9) and Ang-(1-5) formation.[15] Renin is profoundly selective for its only naturally occurring substrate which is angiotensinogen. This means that renin inhibitors that have a high specificity for human renin, theoretically should have very few side effects, if any. This has been shown for aliskiren which has a placebo-like side-effect profile.[16]
Ang II also functions within the RAAS as a negative feedback to suppress further release of renin. This reduction in Ang II levels suppress the feedback loop. That leads to further increased plasma renin concentrations (PRC) and plasma renin activity (PRA). This can be problematic for ACE inhibitor and angiotensin II receptor antagonist therapy since increased PRA could partially overcome the pharmacologic inhibition of the RAAS cascade. Because renin inhibitors directly effect renin activity, decrease of PRA despite the increased PRC (from loss of the negative feedback) may be clinically advantageous.[17]
Drug discovery and development
Pepstatin – the first renin inhibitor
Pepstatin was the first synthetic renin inhibitor. It is of microbial origin and is an N-acyl-pentapeptide, more accurately: isovaleryl-L-valyl-L-valyl-statyl-L-alanyl-statine.[2][18] Pepstatin was found to be a potent competitive inhibitor of most aspartic proteases but a weak inhibitor of renin.[19] Originally it was thought to be effective in the treatment of duodenal ulcer and went through clinical trials but had no success.[20][21] Statine, an amino acid, is thought to be responsible for the inhibitory activity of pepstatin. That is because it mimics the tetrahedral transition state of the peptide catalysis.[22] Because of hydrophobic properties of statine, pepstatin has very low solubility in physiological media.[23] Since it had low potency and poor solubility it did not enter in vivo studies.
First generation – peptide analogues
 H-142[clarification needed]
H-142[clarification needed]This generation consists of two groups of compounds, either peptide analogues of the prosegment of renin[24] or peptide analogues of the amino-terminal part of the substrate angiotensinogen.[7][25][26] The drugs in the latter group seemed to be effective in inhibiting renin activity and lowering blood pressure in both animals and humans.[27] Unfortunately they had to be given parenterally because of poor bioavailability. They also turned out to have a short duration of action, low potency and their ability to lower blood pressure was inadequate. None of these drugs completed clinical investigations.[17]
Second generation – peptide mimetics
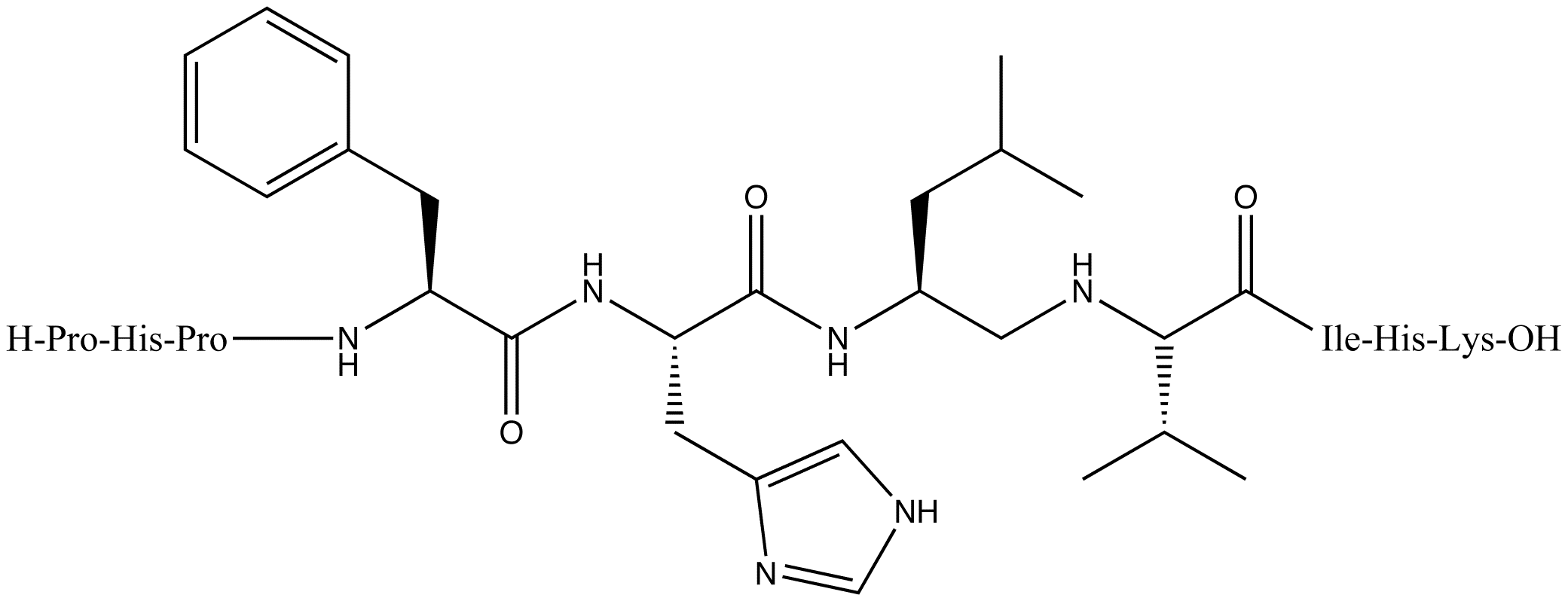
 Remikiren, a second-generation renin inhibitor
Remikiren, a second-generation renin inhibitorCompounds in this generation were more potent, more stable and had longer duration of action. One of these, CGP2928, a peptidomimetic compound was the first renin inhibitor that proved effective when taken orally. It was tested on marmosets and was only active at high doses.[6] Development of new drugs in the second generation continued to improve pharmacokinetic properties. Remikiren, enalkiren and zankiren were then discovered. These were peptidomimetic inhibitors with improved structure that made them more specific, potent and stable. Unfortunately the clinical development was terminated because the drugs had poor oral bioavailability (it was poorly absorbed and rapidly metabolized) and lowering blood pressure activity still remained low including short half-life.[1][13][17]
Third generation – non-peptide mimetics
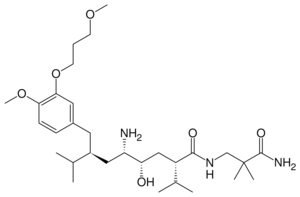
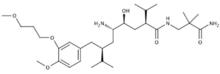 Aliskiren, the third-generation renin inhibitor
Aliskiren, the third-generation renin inhibitorAliskiren is an orally active non-peptide renin inhibitor. It was the first drug in its class on the market. It is used to treat hypertension as monotherapy or in combination with other antihypertensive agents.[1] The key to the discovery of aliskiren was crystallography and molecular modeling techniques. Now a solution has been found to the problem that impeded the development of the renin inhibitors of the previous generations. It was known that non-peptide mimetics would solve the problems of poor pharmacokinetic properties and low specificity. This led to the design of small molecules, nonpeptide inhibitors, which were very potent and specific of human renin.[13][28]
Binding and structure activity relationship of renin inhibitors
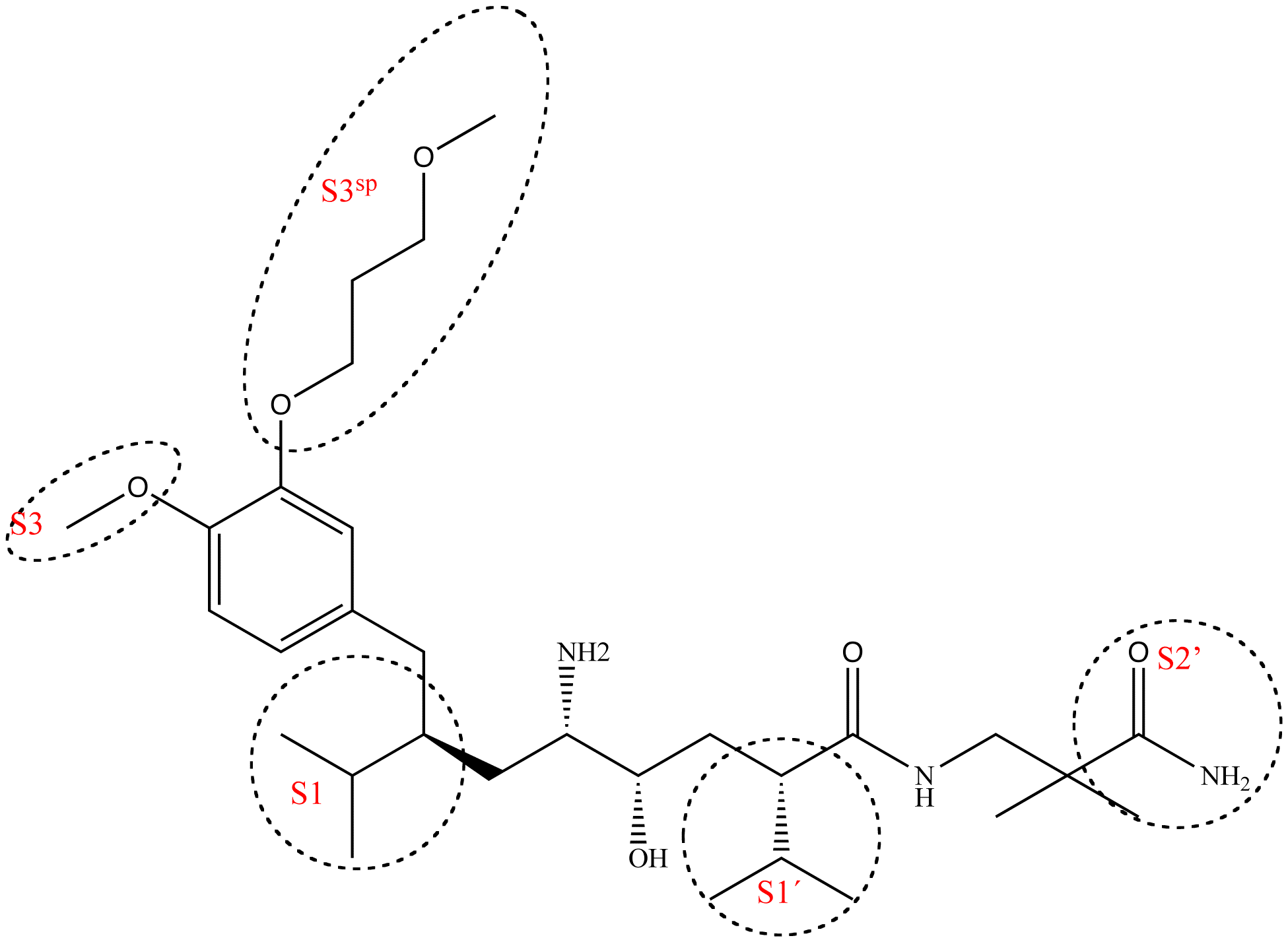
The renin molecule is a monospecific enzyme that belongs to the aspartic protease family.[29] Its structure is complex and consists of two homologous lobes that fold mainly in a β-sheet conformation.[13] Between the two lobes, deep within the enzyme, resides the active site and its catalytic activity is due to two aspartic acid residues (Asp32 and Asp 215, one from each lobe in the renin molecule).[30] A flexible flap made from amino acids formed in a β-hairpin closes the active site by covering the cleft.[31] The renin molecule contains both hydrophobic and hydrophilic amino acids. The hydrophilic tend to be on the outside of the molecule while the hydrophobic tend to be more on the inside and form the active site, a large hydrophobic cavity[32] that can accommodate a ligand with at least seven residues. The principal connection between a ligand and the enzyme is by hydrogen bonding. The residues are named after their place in the ligand, the residues closest to the cleavage site are named P1 and P1′ and they bind into the S1 and S1′ pockets respectively. There are four S pockets, and three S′ pockets, see table 1. The pockets alternate on either side of the backbone in the ligand. This alternation affects the orientation of the pockets making the S3 and S1 pockets arrange together and it makes the S2 pocket close to both S4 and S1′ pockets.[31] Evidence suggest that the closely arranged S1 and S3 pockets merge to form a spacious superpocket.[33] Ligands that fill the superpocket have greater potency than those who do not, occupying increases potency 200 fold. These ligands can be structurally diverse and form van der Waals bonds to the surface of the superpocket.[5] From the S3 pocket stretches a binding site that is distinct for renin, the S3sp subpocket.[29] The S3sp subpocket can accommodate both hydrophobic and polar residues, the pocket can accommodate three water molecules but has also lipophillic nature. The S3sp subpocket is not conformationally flexible so the residues occupying the pocket must have certain characteristics. They can not be sterically demanding and must have reasonably high number of rotatable bonds and be able to connect with hydrogen bonds. The S2 pocket is large, bipartite and hydrophobic but can accommodate both hydrophobic and polar ligands. This diversity of possible polarity offers the P2 residue opportunity of variation in its connection to the enzyme. The S3-S1 and the S3sp subpocket have been the main target of drug design but recent discoveries have indicated that there are other sites of interest. Interactions to the pockets on the S′ site have been proven to be critical for affinity, especially the S1′ and S2′, and in vitro test have indicated that interaction with the flap region could be important to affinity.[5]
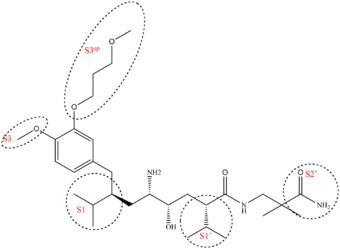 Binding pockets that aliskiren connects with
Binding pockets that aliskiren connects withCharacteristics of each pocket and the importance each residue in the ligand has to binding Pocket Characteristics[5] Subsite Importance to binding[5][34] S4 Hydrophobic P4 Relatively important for binding S3 Hydrophobic P3 Very important for binding S3sp Equally hydrophobic/-philic P3 side chain Dramatically enhances binding affinity S2 Large and hydrophobic P2 Important for binding S1 Large and hydrophobic P1 NA S1′ Primarily hydrophobic P1′ Critical for tight binding S2′ Polar P2′ Critical for tight binding S3′ NA P3′ Structure and presence is not as important Interaction with both aspartic acids in the active site results in a higher affinity. Higher affinity also results by occupying more active site pockets. However some pockets contribute more to the affinity than others, see table 1. A hydrophobic interaction with the S3sp subpocket, S1 and S3 contribute to higher potency and affinity.[35] By having a large and aromatic residue in P3 increases inhibitory activity.[36] Occupation of the S3sp subpocket can increase potency by 50 fold and results in tight binding.[5]
Example of binding to the renin inhibitor: Aliskiren is a peptide like renin inhibitor and unlike most it is rather hydrophilic. It blocks the catalytic function of the enzyme by occupying the S3 to S2′ pockets, except the S2 pocket. Aliskiren also binds to the S3sp subpocket and because that pocket is distinct for renin aliskiren doesn't inhibit other aspartic proteases such as cathepsin D and pepsin.[34] The side chain of aliskiren that binds the S3sp subpocket is ideal and leads to its quality as an inhibitor of human renin.[5] The hydroxyl group in aliskiren forms a hydrogen bond with both oxygen atoms of the Asp32. The amine group forms a hydrogen bond with the carboxylic acid group of Gly217 and the oxygen atom of the Asp32. The methoxy group on the aromatic ring fills the S3 pocket and may possibly form a hydrogen bond with a secondary amine group of Tyr14. The amide group forms a hydrogen bond with a secondary amine group of Ser76.[35] The S1 and S1′ pockets are occupied by the two propyl groups in positions P1 and P1′.[33] The terminal amide in position P2′ anchors the amide tail in the active site by forming a hydrogen bond with Arg74 in the S2′ pocket.[37]
Current status
The first renin inhibitor approved for the treatment of hypertension, aliskiren, has not only shown to be effective in lowering blood pressure but also has fewer side-effects compared with ACE inhibitors. Moreover the renin inhibitor indicates that it provides organ protection, especially for patient with concomitant renal dysfunction and cardiovascular disease.[1][17]
Aliskiren has been in development as a monotherapy and in combination with other high blood pressure medicines. Aliskiren in combination with hydrochlorothiazide was approved by the FDA in 2008 under the tradename Tekturna HCT.[38][39]
In 2007 the companies Actelion/Merck and Speedel announced that they have the next generation of renin inhibitors in clinical research. The lead compound from Actelion/Merck has entered phase II trials. One compound from Speedel, SPP635, has completed phase IIa. The results showed that it was safe and well tolerated over four weeks period and it reduced the blood pressure by 9,8 mmHg – 17,9 mmHg. In 2008 SPP635 was continuing phase II development for hypertension in diabetic patients. More renin inhibitors from Speedel are in clinical trials. Two of them, SPP1148 and SPP676, have entered phase I. Other are in preclinical phases, the compound SPP1234 and compounds from the SPP800 series.[38]
The next generation of renin inhibitors have shown potential improvements over previous generations were bioavailability has increased up to 30% in humans, and they have better tissue distribution.[38][unreliable source]
See also
- ACE inhibitor
- ACE inhibitors drug design
- Aliskiren
- Angiotensin
- Angiotensin II receptor antagonist
- Beta blocker
- Circulatory system
- Discovery and development of angiotensin receptor blockers
- Renin-angiotensin system
- Renin inhibitor
References
- ^ a b c d e f g h i j k Jensen, C.; Herold, P.; Brunner, H. R. (2008). "Aliskiren: The first renin inhibitor for clinical treatment". Nature Reviews Drug Discovery 7 (5): 399–410. doi:10.1038/nrd2550. PMID 18340340.
- ^ a b c Gross, F.; Lazar, J.; Orth, H. (1972). "Inhibition of the renin-angiotensinogen reaction by pepstatin". Science 175 (22): 656. PMID 4109853.
- ^ a b Ferrario, C. M.; Iyer, S. N. (1998). "Angiotensin-(1-7): A bioactive fragment of the renin-angiotensin system". Regulatory peptides 78 (1–3): 13–18. PMID 9879742.
- ^ a b Phillips, M. I.; Schmidt-Ott, K. M. (1999). "The Discovery of Renin 100 Years Ago". News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society 14: 271–274. PMID 11390864.
- ^ a b c d e f g h Webb, R. L.; Schiering, N.; Sedrani, R.; Maibaum, J. R. (2010). "Direct Renin Inhibitors as a New Therapy for Hypertension". Journal of Medicinal Chemistry 53 (21): 7490–7520. doi:10.1021/jm901885s. PMID 20731374.
- ^ a b c Wood, J. M.; Gulati, N.; Forgiarini, P.; Fuhrer, W.; Hofbauer, K. G. (1985). "Effects of a specific and long-acting renin inhibitor in the marmoset". Hypertension 7 (5): 797–803. PMID 3928488.
- ^ a b Szelke, M.; Leckie, B.; Hallett, A.; Jones, D. M.; Sueiras, J.; Atrash, B.; Lever, A. F. (1982). "Potent new inhibitors of human renin". Nature 299 (5883): 555–557. PMID 6750410.
- ^ Segall, L.; Covic, A.; Goldsmith, D. J. A. (2007). "Direct renin inhibitors: The dawn of a new era, or just a variation on a theme?". Nephrology Dialysis Transplantation 22 (9): 2435–2439. doi:10.1093/ndt/gfm363. PMID 17556409.
- ^ Nussberger, J.; Wuerzner, G.; Jensen, C.; Brunner, H. R. (2002). "Angiotensin II suppression in humans by the orally active renin inhibitor Aliskiren (SPP100): Comparison with enalapril". Hypertension 39 (1): E1–E8. PMID 11799102.
- ^ a b c d e Brown, M. J. (2006). Direct renin inhibition — a new way of targeting the renin system. Journal of Renin-Angiotensin-Aldosterone System, 7(2 suppl), S7-S11. http://jra.sagepub.com/content/7/2_suppl/S7.abstract
- ^ Tice, C. M.; Xu, Z.; Yuan, J.; Simpson, R. D.; Cacatian, S. T.; Flaherty, P. T.; Zhao, W.; Guo, J. et al. (2009). "Design and optimization of renin inhibitors: Orally bioavailable alkyl amines". Bioorganic & Medicinal Chemistry Letters 19 (13): 3541–3545. doi:10.1016/j.bmcl.2009.04.140. PMID 19457666.
- ^ a b c Ferrario, C. M. (2006). "Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research". Journal of the renin-angiotensin-aldosterone system : JRAAS 7 (1): 3–14. doi:10.3317/jraas.2006.003. PMID 17083068.
- ^ a b c d Rahuel, J.; Rasetti, V.; Maibaum, J.; Rüeger, H.; Göschke, R.; Cohen, N. C.; Stutz, S.; Cumin, F. et al. (2000). "Structure-based drug design: The discovery of novel nonpeptide orally active inhibitors of human renin". Chemistry & biology 7 (7): 493–504. PMID 10903938.
- ^ a b c Hsueh, W. A.; Wyne, K. (2011). "Renin-Angiotensin-Aldosterone System in Diabetes and Hypertension". The Journal of Clinical Hypertension 13 (4): 224–237. doi:10.1111/j.1751-7176.2011.00449.x. PMID 21466617.
- ^ Müller, D. N.; Derer, W.; Dechend, R. (2008). "Aliskiren—mode of action and preclinical data". Journal of Molecular Medicine 86 (6): 659–662. doi:10.1007/s00109-008-0330-6. PMID 18443751.
- ^ Weir, M.; Bush, C.; Anderson, D.; Zhang, J.; Keefe, D.; Satlin, A. (2007). "Antihypertensive efficacy, safety, and tolerability of the oral direct renin inhibitor aliskiren in patients with hypertension: A pooled analysis". Journal of the American Society of Hypertension 1 (4): 264–277. doi:10.1016/j.jash.2007.04.004. PMID 20409858.
- ^ a b c d Staessen, J. A.; Li, Y.; Richart, T. (2006). "Oral renin inhibitors". The Lancet 368 (9545): 1449–1456. doi:10.1016/S0140-6736(06)69442-7. PMID 17055947.
- ^ Umezawa, H.; Aoyagi, T.; Morishima, H.; Matsuzaki, M.; Hamada, M. (1970). "Pepstatin, a new pepsin inhibitor produced by Actinomycetes". The Journal of antibiotics 23 (5): 259–262. PMID 4912600.
- ^ Fisher, N. D. L.; Hollenberg, N. K. (2005). "Renin Inhibition: What Are the Therapeutic Opportunities?". Journal of the American Society of Nephrology 16 (3): 592–599. doi:10.1681/ASN.2004100874. PMID 15703270.
- ^ Bonnevie, O.; Svendsen, L. B.; Holst-Christensen, J.; Johansen, T. S.; Søltoft, J.; Christiansen, P. M. (1979). "Double-blind randomised clinical trial of a pepsin-inhibitory pentapeptide (pepstatin) in the treatment of duodenal ulcer". Gut 20 (7): 624–628. PMC 1412504. PMID 385457. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1412504.
- ^ Svendsen, L. B.; Christiansen, P. M.; Bonnevie, O. (1979). "Gastric ulcer therapy with a pepsin-inactivating peptide, pepstatin: A double-blind randomized clinical trial". Scandinavian journal of gastroenterology 14 (8): 929–932. PMID 394302.
- ^ Marciniszyn Jr, J.; Hartsuck, J. A.; Tang, J. (1976). "Mode of inhibition of acid proteases by pepstatin". The Journal of biological chemistry 251 (22): 7088–7094. PMID 993206.
- ^ Eid, M.; Evin, G.; Castro, B.; Menard, J.; Corvol, P. (1981). "New renin inhibitors homologous with pepstatin". The Biochemical journal 197 (2): 465–471. PMC 1163147. PMID 7034718. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1163147.
- ^ Cumin, F., Evin, G., Fehrentz, J. A., Seyer, R., Castro, B., Menard, J., et al. (1985). Inhibition of human renin by synthetic peptides derived from its prosegment. J Biol Chem, 260(16), 9154-9157. http://www.jbc.org/content/260/16/9154.abstract
- ^ Szelke, M., Leckie, B. J., Tree, M., Brown, A., Grant, J., Hallett, A., et al. (1982). H-77: a potent new renin inhibitor. In vitro and in vivo studies. Hypertension, 4(3 Pt 2), 59-69. http://hyper.ahajournals.org/content/4/3/59.abstract
- ^ Tree, M.; Atrash, B.; Donovan, B.; Gamble, J.; Hallett, A.; Hughes, M.; Jones, D. M.; Leckie, B. et al. (1983). "New inhibitors of human renin tested in vitro and in vivo in the anaesthetized baboon". Journal of hypertension 1 (4): 399–403. doi:10.1097/00004872-198312000-00013. PMID 6398331.
- ^ Webb, D. J.; Manhem, P. J.; Ball, S. G.; Inglis, G.; Leckie, B. J.; Lever, A. F.; Morton, J. J.; Robertson, J. I. et al. (1985). "A study of the renin inhibitor H142 in man". Journal of hypertension 3 (6): 653–658. PMID 3910726.
- ^ Claude Cohen, N. (2007). "Structure-Based Drug Design and the Discovery of Aliskiren (Tekturna): Perseverance and Creativity to Overcome a R&D Pipeline Challenge". Chemical Biology & Drug Design 70 (6): 557–565. doi:10.1111/j.1747-0285.2007.00599.x. PMID 17999663.
- ^ a b Winiecka, I.; Dudkiewicz-Wilczyńska, J.; Roman, I.; Paruszewski, R. (2010). "New potential renin inhibitors with dipeptide replacements in the molecule". Acta poloniae pharmaceutica 67 (4): 367–374. PMID 20635532.
- ^ Gradman, A. H., & Kad, R. (2008). Renin inhibition in hypertension. [Review]. J Am Coll Cardiol, 51(5), 519-528. http://content.onlinejacc.org/cgi/content/full/51/5/519
- ^ a b Lunney, E. A.; Hamilton, H. W.; Hodges, J. C.; Kaltenbronn, J. S.; Repine, J. T.; Badasso, M.; Cooper, J. B.; Dealwis, C. et al. (1993). "Analyses of ligand binding in five endothiapepsin crystal complexes and their use in the design and evaluation of novel renin inhibitors". Journal of medicinal chemistry 36 (24): 3809–3820. PMID 8254610.
- ^ Matter, H.; Scheiper, B.; Steinhagen, H.; Böcskei, Z.; Fleury, V. R.; McCort, G. (2011). "Structure-based design and optimization of potent renin inhibitors on 5- or 7-azaindole-scaffolds". Bioorganic & Medicinal Chemistry Letters 21 (18): 5487–5492. doi:10.1016/j.bmcl.2011.06.112. PMID 21840215.
- ^ a b Yuan, J.; Simpson, R. D.; Zhao, W.; Tice, C. M.; Xu, Z.; Cacatian, S.; Jia, L.; Flaherty, P. T. et al. (2011). "Biphenyl/diphenyl ether renin inhibitors: Filling the S1 pocket of renin via the S3 pocket". Bioorganic & Medicinal Chemistry Letters 21 (16): 4836–4843. doi:10.1016/j.bmcl.2011.06.043. PMID 21741239.
- ^ a b Wood, J. M.; Maibaum, J.; Rahuel, J.; Grütter, M. G.; Cohen, N. C.; Rasetti, V.; Rüger, H.; Göschke, R. et al. (2003). "Structure-based design of aliskiren, a novel orally effective renin inhibitor". Biochemical and biophysical research communications 308 (4): 698–705. PMID 12927775.
- ^ a b Politi, A.; Durdagi, S.; Moutevelis-Minakakis, P.; Kokotos, G.; Mavromoustakos, T. (2010). "Development of accurate binding affinity predictions of novel renin inhibitors through molecular docking studies". Journal of Molecular Graphics and Modelling 29 (3): 425–435. doi:10.1016/j.jmgm.2010.08.003. PMID 20855222.
- ^ Akahane, K.; Umeyama, H.; Nakagawa, S.; Moriguchi, I.; Hirose, S.; Iizuka, K.; Murakami, K. (1985). "Three-dimensional structure of human renin". Hypertension 7 (1): 3–12. PMID 3884499.
- ^ Wu, Y.; Shi, C.; Sun, X.; Wu, X.; Sun, H. (2011). "Synthesis, biological evaluation and docking studies of octane-carboxamide based renin inhibitors with extended segments toward S3′ site of renin". Bioorganic & Medicinal Chemistry 19 (14): 4238–4249. doi:10.1016/j.bmc.2011.05.059. PMID 21708467.
- ^ a b c Speedel Acquiring an additional 51.7% stake and announcing plans for mandatory public tender offer.TRANSACTION OVERVIEW. (2008). From Novartis: http://www.novartis.com/downloads/investors/presentations-events/other-events/2008/2008-07_speedel-backgrounder.pdf
- ^ Tekturna HCT (aliskiren; hydrochlorothiazide) tablets. (2011). From US Food and Drug Administration: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022107s009lbl.pdf
Categories:- Renin inhibitors
- Drug discovery
Wikimedia Foundation. 2010.