- DNA sequencing
-
Part of a series on Genetics Key components Chromosome
DNA • RNA
Genome
Heredity
Mutation
Nucleotide
VariationHistory and topics Evolution • Molecular
Population genetics
Mendelian inheritance
Quantitative genetics
Molecular geneticsResearch DNA sequencing
Genetic engineering
Genomics • Topics
Medical geneticsBiology portal • DNA sequencing includes several methods and technologies that are used for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA.
Knowledge of DNA sequences has become indispensable for basic biological research, other research branches utilizing DNA sequencing, and in numerous applied fields such as diagnostic, biotechnology, forensic biology and biological systematics. The advent of DNA sequencing has significantly accelerated biological research and discovery. The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the sequencing of the human genome, in the Human Genome Project. Related projects, often by scientific collaboration across continents, have generated the complete DNA sequences of many animal, plant, and microbial genomes.
The first DNA sequences were obtained in the early 1970s by academic researchers using laborious methods based on two-dimensional chromatography. Following the development of dye-based sequencing methods with automated analysis,[1] DNA sequencing has become easier and orders of magnitude faster.[2]
History
RNA sequencing was one of the earliest forms of nucleotide sequencing. The major landmark of RNA sequencing is the sequence of the first complete gene and the complete genome of Bacteriophage MS2, identified and published by Walter Fiers and his coworkers at the University of Ghent (Ghent, Belgium), between 1972[3] and 1976.[4]
Prior to the development of rapid DNA sequencing methods in the early 1970s by Frederick Sanger at the University of Cambridge, in England and Walter Gilbert and Allan Maxam at Harvard,[5][6] a number of laborious methods were used. For instance, in 1973, Gilbert and Maxam reported the sequence of 24 basepairs using a method known as wandering-spot analysis.[7]
The chain-termination method developed by Sanger and coworkers in 1977 soon became the method of choice, owing to its relative ease and reliability.[8][9] It involves separating DNA bases from different DNA fragments.[citation needed]
Maxam–Gilbert sequencing
In 1976–1977, Allan Maxam and Walter Gilbert developed a DNA sequencing method based on chemical modification of DNA and subsequent cleavage at specific bases.[5] Although Maxam and Gilbert published their chemical sequencing method two years after the ground-breaking paper of Sanger and Coulson on plus-minus sequencing,[8][10] Maxam–Gilbert sequencing rapidly became more popular, since purified DNA could be used directly, while the initial Sanger method required that each read start be cloned for production of single-stranded DNA. However, with the improvement of the chain-termination method (see below), Maxam-Gilbert sequencing has fallen out of favour due to its technical complexity prohibiting its use in standard molecular biology kits, extensive use of hazardous chemicals, and difficulties with scale-up.[11]
The method requires radioactive labeling at one 5' end of the DNA (typically by a kinase reaction using gamma-32P ATP) and purification of the DNA fragment to be sequenced. Chemical treatment generates breaks at a small proportion of one or two of the four nucleotide bases in each of four reactions (G, A+G, C, C+T). For example, the purines (A+G) are depurinated using formic acid, the guanines (and to some extent the adenines) are methylated by dimethyl sulfate, and the pyrimidines (C+T) are methylated using hydrazine. The addition of salt (sodium chloride) to the hydrazine reaction inhibits the methylation of thymine for the C-only reaction. The modified DNAs are then cleaved by hot piperidine at the position of the modified base. The concentration of the modifying chemicals is controlled to introduce on average one modification per DNA molecule. Thus a series of labeled fragments is generated, from the radiolabeled end to the first "cut" site in each molecule. The fragments in the four reactions are electrophoresed side by side in denaturing acrylamide gels for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each corresponding to a radiolabeled DNA fragment, from which the sequence may be inferred.[12]
Also sometimes known as "chemical sequencing", this method led to the Methylation Interference Assay used to map DNA-binding sites for DNA-binding proteins.[13]
Chain-termination methods
 Part of a radioactively labelled sequencing gel
Part of a radioactively labelled sequencing gel
Because the chain-terminator method (or Sanger method after its developer Frederick Sanger) is more efficient and uses fewer toxic chemicals and lower amounts of radioactivity than the method of Maxam and Gilbert, it rapidly became the method of choice. The key principle of the Sanger method was the use of dideoxynucleotide triphosphates (ddNTPs) as DNA chain terminators.
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotidetriphosphates (dNTPs), and modified nucleotides (dideoxyNTPs) that terminate DNA strand elongation. These ddNTPs will also be radioactively or fluorescently labelled for detection in automated sequencing machines. The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP) which are the chain-terminating nucleotides, lacking a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, thus terminating DNA strand extension and resulting in DNA fragments of varying length.
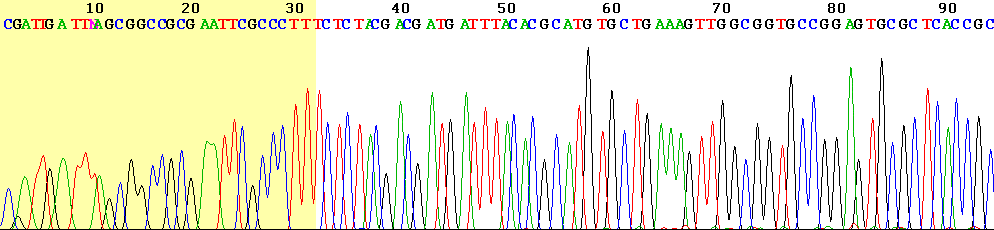
The newly synthesized and labelled DNA fragments are heat denatured, and separated by size (with a resolution of just one nucleotide) by gel electrophoresis on a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C); the DNA bands are then visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image. In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes are then used to read (from bottom to top) the DNA sequence.
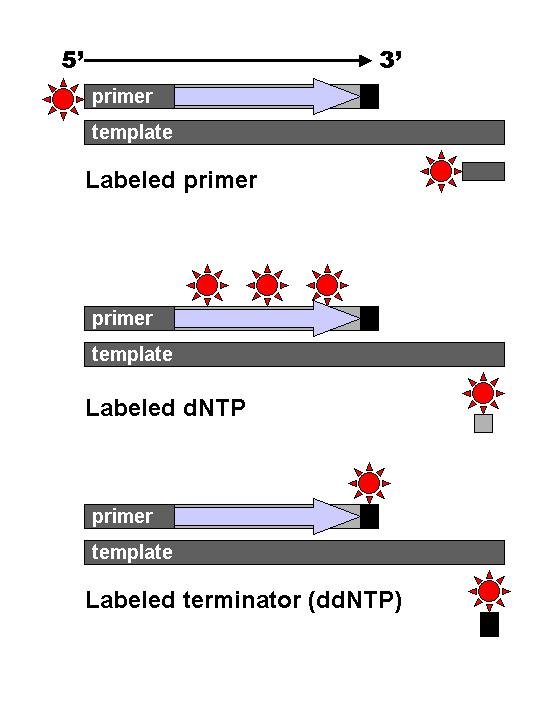
 DNA fragments are labelled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP. (click to expand)
DNA fragments are labelled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP. (click to expand)Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5' end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers [14][15] of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
 Sequence ladder by radioactive sequencing compared to fluorescent peaks (click to expand)
Sequence ladder by radioactive sequencing compared to fluorescent peaks (click to expand)Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
Dye-terminator sequencing
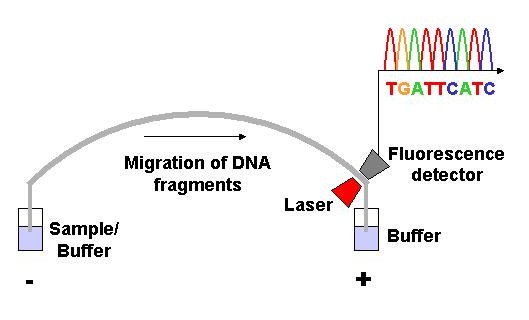
 Capillary electrophoresis (click to expand)
Capillary electrophoresis (click to expand)Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction, rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emit light at different wavelengths.
Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Its limitations include dye effects due to differences in the incorporation of the dye-labelled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace chromatogram after capillary electrophoresis (see figure to the left).
This problem has been addressed with the use of modified DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating "dye blobs". The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, is now being used for the vast majority of sequencing projects.
Challenges
Common challenges of DNA sequencing include poor quality in the first 15–40 bases of the sequence and deteriorating quality of sequencing traces after 700–900 bases. Base calling software typically gives an estimate of quality to aid in quality trimming.[16][17]
In cases where DNA fragments are cloned before sequencing, the resulting sequence may contain parts of the cloning vector. In contrast, PCR-based cloning and emerging sequencing technologies based on pyrosequencing often avoid using cloning vectors. Recently, one-step Sanger sequencing (combined amplification and sequencing) methods such as Ampliseq and SeqSharp have been developed that allow rapid sequencing of target genes without cloning or prior amplification.[18][19]
Current methods can directly sequence only relatively short (300–1000 nucleotides long) DNA fragments in a single reaction. The main obstacle to sequencing DNA fragments above this size limit is insufficient power of separation for resolving large DNA fragments that differ in length by only one nucleotide. In all cases the use of a primer with a free 3' end is essential.
Automation and sample preparation
 View of the start of an example dye-terminator read (click to expand)
View of the start of an example dye-terminator read (click to expand)Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch (run) in up to 24 runs a day. DNA sequencers carry out capillary electrophoresis for size separation, detection and recording of dye fluorescence, and data output as fluorescent peak trace chromatograms. Sequencing reactions by thermocycling, cleanup and re-suspension in a buffer solution before loading onto the sequencer are performed separately. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (generally located at the ends of the sequence). The accuracy of such algorithms is below visual examination by a human operator, but sufficient for automated processing of large sequence data sets.
Amplification and clonal selection
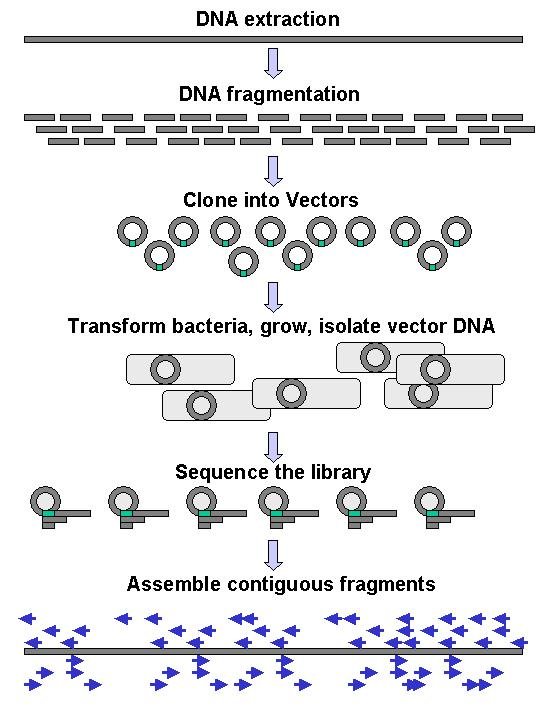
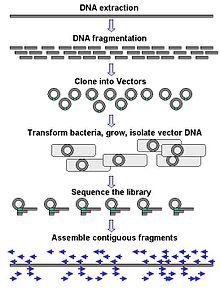 Genomic DNA is fragmented into random pieces and cloned as a bacterial library. DNA from individual bacterial clones is sequenced and the sequence is assembled by using overlapping DNA regions.(click to expand)
Genomic DNA is fragmented into random pieces and cloned as a bacterial library. DNA from individual bacterial clones is sequenced and the sequence is assembled by using overlapping DNA regions.(click to expand)Large-scale sequencing often aims at sequencing very long DNA pieces, such as whole chromosomes, although large scale sequencing can also be used to generate very large numbers of short sequences, such as found in phage display. For longer targets, such as chromosomes, common approaches consist of cutting (with restriction enzymes) or shearing (with mechanical forces) large DNA fragments into shorter DNA fragments. The fragmented DNA is cloned into a DNA vector, and amplified in Escherichia coli. Short DNA fragments purified from individual bacterial colonies are individually sequenced and assembled electronically into one long, contiguous sequence.
This method does not require any pre-existing information about the sequence of the DNA and is referred to as de novo sequencing. Gaps in the assembled sequence may be filled by primer walking. The different strategies have different tradeoffs in speed and accuracy; shotgun methods are often used for sequencing large genomes, but its assembly is complex and difficult, particularly with sequence repeats often causing gaps in genome assembly.
Most sequencing approaches use an in vitro cloning step to amplify individual DNA molecules, because their molecular detection methods are not sensitive enough for single molecule sequencing. Emulsion PCR[20] isolates individual DNA molecules along with primer-coated beads in aqueous droplets within an oil phase. Polymerase chain reaction (PCR) then coats each bead with clonal copies of the DNA molecule followed by immobilization for later sequencing. Emulsion PCR is used in the methods by Marguilis et al. (commercialized by 454 Life Sciences), Shendure and Porreca et al. (also known as "Polony sequencing") and SOLiD sequencing, (developed by Agencourt, now Applied Biosystems).[21][22][23]
Another method for in vitro clonal amplification is bridge PCR, where fragments are amplified upon primers attached to a solid surface, used in the Illumina Genome Analyzer. Single-molecule methods, such as that developed by Stephen Quake's laboratory (later commercialized by Helicos) is an exception: it uses bright fluorophores and laser excitation to detect base addition events from individual DNA molecules fixed to a surface, eliminating the need for molecular amplification.[24]
High-throughput sequencing
The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.[25][26] High-throughput sequencing technologies are intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods.[27]
Lynx Therapeutics' Massively Parallel Signature Sequencing (MPSS)
Main article: Massively parallel signature sequencingThe first of the "next-generation" sequencing technologies, MPSS was developed in the 1990s at Lynx Therapeutics, a company founded in 1992 by Sydney Brenner and Sam Eletr. MPSS was a bead-based method that used a complex approach of adapter ligation followed by adapter decoding, reading the sequence in increments of four nucleotides; this method made it susceptible to sequence-specific bias or loss of specific sequences. Because the technology was so complex, MPSS was only performed 'in-house' by Lynx Therapeutics and no machines were sold; when the merger with Solexa later led to the development of sequencing-by-synthesis, a more simple approach with numerous advantages, MPSS became obsolete. However, the essential properties of the MPSS output were typical of later "next-gen" data types, including hundreds of thousands of short DNA sequences. In the case of MPSS, these were typically used for sequencing cDNA for measurements of gene expression levels. Lynx Therapeutics merged with Solexa in 2004, and this company was later purchased by Illumina. [28]
Polony sequencing
Polony sequencing, developed in the laboratory of George Church at Harvard, was among the first next-generation sequencing systems used to sequence a full genome in 2005. It combined an in vitro paired-tag library with emulsion PCR, an automated microscope, and ligation-based sequencing chemistry to sequence an E. coli genome at an accuracy of > 99.9999% and a cost approximately 1/10 that of Sanger sequencing. The technology was licensed to Agencourt Biosciences, subsequently spun out into Agencourt Personal Genomics, and ultimately incorporated into the Applied Biosystems SOLiD platform.
454 pyrosequencing
Main article: 454 Life Sciences#TechnologyA parallelized version of pyrosequencing was developed by 454 Life Sciences, which has since been acquired by Roche Diagnostics. The method amplifies DNA inside water droplets in an oil solution (emulsion PCR), with each droplet containing a single DNA template attached to a single primer-coated bead that then forms a clonal colony. The sequencing machine contains many picolitre-volume wells each containing a single bead and sequencing enzymes. Pyrosequencing uses luciferase to generate light for detection of the individual nucleotides added to the nascent DNA, and the combined data are used to generate sequence read-outs.[21] This technology provides intermediate read length and price per base compared to Sanger sequencing on one end and Solexa and SOLiD on the other.[29]
Illumina (Solexa) sequencing
Solexa, now part of Illumina, developed a sequencing technology based on reversible dye-terminators. DNA molecules are first attached to primers on a slide and amplified so that local clonal colonies are formed (bridge amplification). Four types of ddNTPs are added, and non-incorporated nucleotides are washed away. Unlike pyrosequencing, the DNA can only be extended one nucleotide at a time. A camera takes images of the fluorescently labeled nucleotides, then the dye along with the terminal 3' blocker is chemically removed from the DNA, allowing the next cycle.[30]
SOLiD sequencing
Main article: ABI Solid SequencingApplied Biosystems' SOLiD technology employs sequencing by ligation. Here, a pool of all possible oligonucleotides of a fixed length are labeled according to the sequenced position. Oligonucleotides are annealed and ligated; the preferential ligation by DNA ligase for matching sequences results in a signal informative of the nucleotide at that position. Before sequencing, the DNA is amplified by emulsion PCR. The resulting bead, each containing only copies of the same DNA molecule, are deposited on a glass slide.[31] The result is sequences of quantities and lengths comparable to Illumina sequencing.[29]
Ion semiconductor sequencing
Main article: Ion semiconductor sequencingIon Torrent Systems Inc. developed a system based on using standard sequencing chemistry, but with a novel, semiconductor based detection system. This method of sequencing is based on the detection of hydrogen ions that are released during the polymerisation of DNA, as opposed to the optical methods used in other sequencing systems. A microwell containing a template DNA strand to be sequenced is flooded with a single type of nucleotide. If the introduced nucleotide is complementary to the leading template nucleotide it is incorporated into the growing complementary strand. This causes the release of a hydrogen ion that triggers a hypersensitive ion sensor, which indicates that a reaction has occurred. If homopolymer repeats are present in the template sequence multiple nucleotides will be incorporated in a single cycle. This leads to a corresponding number of released hydrogens and a proportionally higher electronic signal.[32]
DNA nanoball sequencing
Main article: DNA nanoball sequencingDNA nanoball sequencing is a type of high throughput sequencing technology used to determine the entire genomic sequence of an organism. The company Complete Genomics uses this technology to sequence samples that researchers submit from several projects. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Unchained sequencing by ligation is then used to determine the nucleotide sequence.[33] This method of DNA sequencing allows large numbers of DNA nanoballs to be sequenced per run and at low reagent costs compared to other next generation sequencing platforms.[34] However, only short sequences of DNA are determined from each DNA nanoball which makes mapping the short reads to a reference genome difficult.[35] This technology has been used for multiple genome sequencing projects and is scheduled to be used for more.[36]
Helioscope(TM) single molecule sequencing
Main article: Helioscope (TM) single molecule sequencingBased on "true single molecule sequencing" technology, Helioscope sequencing uses DNA fragments with added polyA tail adapters, which are attached to the flow cell surface. The next steps involve extension-based sequencing with cyclic washes of the flow cell with fluorescently labeled nucleotides (one nucleotide type at a time, as with the Sanger method). The reads are performed by the Helioscope sequencer. The reads are short, up to 55 bases per run, but recent improvemend of the methodology allowes more accurate reads of homopolymers (stretches of one type of nucleotides) and RNA sequencing.
Single Molecule SMRT(TM) sequencing
Main article: Single molecule SMRT (TM) sequencingSMRT sequencing is based on the sequencing by synthesis approach. The DNA is synthesisd in so calles zero-mode wave-guides (ZMWs) - small well-like containers with the capturing tools located at the bottom of the well. The sequencing is performed with use of unmodified polymerase (attached to the ZMW bottom) and fluorescently labelled nucleotides flowing freely in the solution. The wells are constructed in a way that only the fluorescence occurring by the bottom of the well is detected. The fluorescent label is detached from the nucleotide at its incorporation into the DNA strand, leaving an unmodified DNA strand. According to Pacific Biosciences, the SMTR technology developer, this methodology allows detection of nucleotide modifications (such ad cytosine methylation). This happens through the observation of polymerase kinetics. This approach allows reads of 1000 nucleotides.
Single Molecule real time (RNAP) sequencing
Main article: Single Molecule real time (RNAP)sequencingThis method is based on RNA polymerase (RNAP), which is attached to a polystyrene bead, with distal end of sequenced DNA is attached to another bead, with both beads being placed in optical traps. RNAP motion during transcription brings the beads in closer and their relative distance changes, which can then be recorded at a single nucleotide resolution. The sequence is deduced based on the four readouts with lowered concentrations of each of the four nucleotide types (similarly to Sangers method).
Nanopore DNA sequencing
Main article: Nanopore sequencingThis method is based on the readout of electrical signal occurring at nucleotides passing by alpha-hemolysin pores covalently bound with cyclodextrin. The DNA passing through the nanopore changes its ion current. This change is dependent on the shape, size and length of the DNA sequence. Each type of the nucleotide blocks the ion flow through the pore for a different period of time. The method has a potential of development as it does not require modified nucleotides, however single nucleotide resolution is not yet available.
VisiGen Biotechnologies approach
Main article: VisiGen Biotechnologies approachVisiGen Biotechnologies introduced a specially engineered DNA polymerase for use in their sequencing. This polymerase acts as a sensor - having incorporated a donor fluorescent dye by its active centre. This donor dye acts by FRET (fluorescent resonant energy transfer), inducing fluorescence of differently labeled nucleotides. This approach allows reads performed at the spead at which polymerase incorporates nucleotides into the sequence (several hundred per second). The nucleotide fluorochrome is released after the incorporation into the DNA strand. The expected read lengths in this approach should reach 1000 nucleotides, however this will have to be confirmed.
Future methods
Sequencing by hybridization is a non-enzymatic method that uses a DNA microarray. A single pool of DNA whose sequence is to be determined is fluorescently labeled and hybridized to an array containing known sequences. Strong hybridization signals from a given spot on the array identifies its sequence in the DNA being sequenced.[37] Mass spectrometry may be used to determine mass differences between DNA fragments produced in chain-termination reactions.[38]
DNA sequencing methods currently under development include labeling the DNA polymerase,[39] reading the sequence as a DNA strand transits through nanopores,[40][41] and microscopy-based techniques, such as AFM or transmission electron microscopy that are used to identify the positions of individual nucleotides within long DNA fragments (>5,000 bp) by nucleotide labeling with heavier elements (e.g., halogens) for visual detection and recording.[42][43] Third generation technologies aim to increase throughput and decrease the time to result and cost by eliminating the need for excessive reagents and harnessing the processivity of DNA polymerase.[44]
In microfluidic Sanger sequencing the entire thermocycling amplification of DNA fragments as well as their separation by electrophoresis is done on a single glass wafer (approximately 10 cm in diameter) thus reducing the reagent usage as well as cost.[citation needed] In some instances researchers have shown that they can increase the throughput of conventional sequencing through the use of microchips.[45] Research will still need to be done in order to make this use of technology effective.
In October 2006, the X Prize Foundation established an initiative to promote the development of full genome sequencing technologies, called the Archon X Prize, intending to award $10 million to "the first Team that can build a device and use it to sequence 100 human genomes within 10 days or less, with an accuracy of no more than one error in every 100,000 bases sequenced, with sequences accurately covering at least 98% of the genome, and at a recurring cost of no more than $10,000 (US) per genome."[46]
Each year NHGRI promotes grants for new research and developments in genomics. 2010 grants and 2011 candidates include continuing work in microfluidic, polony and base-heavy sequencing methodologies [47]
Major landmarks in DNA sequencing
- 1953 Discovery of the structure of the DNA double helix.[48]
- 1972 Development of recombinant DNA technology, which permits isolation of defined fragments of DNA; prior to this, the only accessible samples for sequencing were from bacteriophage or virus DNA.
- 1977 The first complete DNA genome to be sequenced is that of bacteriophage φX174.[49]
- 1977 Allan Maxam and Walter Gilbert publish "DNA sequencing by chemical degradation".[5] Frederick Sanger, independently, publishes "DNA sequencing with chain-terminating inhibitors".[9]
- 1984 Medical Research Council scientists decipher the complete DNA sequence of the Epstein-Barr virus, 170 kb.
- 1986 Leroy E. Hood's laboratory at the California Institute of Technology and Smith announce the first semi-automated DNA sequencing machine.
- 1987 Applied Biosystems markets first automated sequencing machine, the model ABI 370.
- 1990 The U.S. National Institutes of Health (NIH) begins large-scale sequencing trials on Mycoplasma capricolum, Escherichia coli, Caenorhabditis elegans, and Saccharomyces cerevisiae (at US$0.75/base).
- 1991 Sequencing of human expressed sequence tags begins in Craig Venter's lab, an attempt to capture the coding fraction of the human genome.[50]
- 1995 Craig Venter, Hamilton Smith, and colleagues at The Institute for Genomic Research (TIGR) publish the first complete genome of a free-living organism, the bacterium Haemophilus influenzae. The circular chromosome contains 1,830,137 bases and its publication in the journal Science[51] marks the first use of whole-genome shotgun sequencing, eliminating the need for initial mapping efforts.
- 1996 Pål Nyrén and his student Mostafa Ronaghi at the Royal Institute of Technology in Stockholm publish their method of pyrosequencing[52]
- 1998 Phil Green and Brent Ewing of the University of Washington publish "
phred" for sequencer data analysis.[53] - 2000 Lynx Therapeutics publishes and markets "MPSS" - a parallelized, adapter/ligation-mediated, bead-based sequencing technology, launching "next-generation" sequencing.[54]
- 2001 A draft sequence of the human genome is published.[55][56]
- 2004 454 Life Sciences markets a parallelized version of pyrosequencing.[57][58] The first version of their machine reduced sequencing costs 6-fold compared to automated Sanger sequencing, and was the second of a new generation of sequencing technologies, after MPSS.[29]
See also
- Cancer genome sequencing
- Complete Genomics
- DNA field-effect transistor
- DNA sequencing theory
- Genome project
- Multiplex ligation-dependent probe amplification
- Sequence profiling tool
- Single Molecule Real Time Sequencing
References
- ^ Olsvik O, Wahlberg J, Petterson B, et al. (January 1993). "Use of automated sequencing of polymerase chain reaction-generated amplicons to identify three types of cholera toxin subunit B in Vibrio cholerae O1 strains". J. Clin. Microbiol. 31 (1): 22–5. PMC 262614. PMID 7678018. http://jcm.asm.org/cgi/pmidlookup?view=long&pmid=7678018.
- ^ Pettersson E, Lundeberg J, Ahmadian A (February 2009). "Generations of sequencing technologies". Genomics 93 (2): 105–11. doi:10.1016/j.ygeno.2008.10.003. PMID 18992322.
- ^ Min Jou W, Haegeman G, Ysebaert M, Fiers W (May 1972). "Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein". Nature 237 (5350): 82–8. Bibcode 1972Natur.237...82J. doi:10.1038/237082a0. PMID 4555447.
- ^ Fiers W, Contreras R, Duerinck F, et al (April 1976). "Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene". Nature 260 (5551): 500–7. Bibcode 1976Natur.260..500F. doi:10.1038/260500a0. PMID 1264203.
- ^ a b c Maxam AM, Gilbert W (February 1977). "A new method for sequencing DNA". Proc. Natl. Acad. Sci. U.S.A. 74 (2): 560–4. Bibcode 1977PNAS...74..560M. doi:10.1073/pnas.74.2.560. PMC 392330. PMID 265521. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=392330.
- ^ Gilbert, W. DNA sequencing and gene structure. Nobel lecture, 8 December 1980.
- ^ Gilbert W, Maxam A (December 1973). "The Nucleotide Sequence of the lac Operator". Proc. Natl. Acad. Sci. U.S.A. 70 (12): 3581–4. Bibcode 1973PNAS...70.3581G. doi:10.1073/pnas.70.12.3581. PMC 427284. PMID 4587255. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=427284.
- ^ a b Sanger F, Coulson AR (May 1975). "A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase". J. Mol. Biol. 94 (3): 441–8. doi:10.1016/0022-2836(75)90213-2. PMID 1100841.
- ^ a b Sanger F, Nicklen S, Coulson AR (December 1977). "DNA sequencing with chain-terminating inhibitors". Proc. Natl. Acad. Sci. U.S.A. 74 (12): 5463–7. Bibcode 1977PNAS...74.5463S. doi:10.1073/pnas.74.12.5463. PMC 431765. PMID 271968. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=431765.
- ^ Sanger F. Determination of nucleotide sequences in DNA. Nobel lecture, 8 December 1980.
- ^ Graziano Pesole; Cecilia Saccone (2003). Handbook of comparative genomics: principles and methodology. New York: Wiley-Liss. pp. 133. ISBN 0-471-39128-X. http://books.google.com/books?id=dXk0JvN2Y-IC&pg=PA133.
- ^ "Cold Spring Harb Protoc -- Sign In Page". http://cshprotocols.cshlp.org/cgi/content/full/2006/1/pdb.prot3804?ijkey=17dc0499c9e0aa3b22887d5332698df00217774f&keytype2=tf_ipsecsha.
- ^ "Cold Spring Harb Protoc -- Sign In Page". http://cshprotocols.cshlp.org/cgi/content/full/2007/12/pdb.prot4812.
- ^ Smith LM, Sanders JZ, Kaiser RJ, et al (1986). "Fluorescence detection in automated DNA sequence analysis". Nature 321 (6071): 674–9. Bibcode 1986Natur.321..674S. doi:10.1038/321674a0. PMID 3713851. "We have developed a method for the partial automation of DNA sequence analysis. Fluorescence detection of the DNA fragments is accomplished by means of a fluorophore covalently attached to the oligonucleotide primer used in enzymatic DNA sequence analysis. A different coloured fluorophore is used for each of the reactions specific for the bases A, C, G and T. The reaction mixtures are combined and co-electrophoresed down a single polyacrylamide gel tube, the separated fluorescent bands of DNA are detected near the bottom of the tube, and the sequence information is acquired directly by computer."
- ^ Smith LM, Fung S, Hunkapiller MW, Hunkapiller TJ, Hood LE (April 1985). "The synthesis of oligonucleotides containing an aliphatic amino group at the 5' terminus: synthesis of fluorescent DNA primers for use in DNA sequence analysis". Nucleic Acids Res. 13 (7): 2399–412. doi:10.1093/nar/13.7.2399. PMC 341163. PMID 4000959. http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=4000959.
- ^ "Phred - Quality Base Calling". http://www.phrap.com/phred/. Retrieved 2011-02-24.
- ^ "Base-calling for next-generation sequencing platforms — Brief Bioinform". http://bib.oxfordjournals.org/content/early/2011/01/18/bib.bbq077.full. Retrieved 2011-02-24.
- ^ Murphy, K.; Berg, K.; Eshleman, J. (2005). "Sequencing of genomic DNA by combined amplification and cycle sequencing reaction". Clinical chemistry 51 (1): 35–39. doi:10.1373/clinchem.2004.039164. PMID 15514094.
- ^ Sengupta, D. .; Cookson, B. . (2010). "SeqSharp: A general approach for improving cycle-sequencing that facilitates a robust one-step combined amplification and sequencing method". The Journal of molecular diagnostics : JMD 12 (3): 272–277. doi:10.2353/jmoldx.2010.090134. PMC 2860461. PMID 20203000. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2860461.
- ^ Richard Williams, Sergio G Peisajovich, Oliver J Miller, Shlomo Magdassi, Dan S Tawfik, Andrew D Griffiths (2006). "Amplification of complex gene libraries by emulsion PCR". Nature methods 3 (7): 545–550. doi:10.1038/nmeth896. PMID 16791213.
- ^ a b Margulies M, Egholm M, Altman WE, et al (September 2005). "Genome Sequencing in Open Microfabricated High Density Picoliter Reactors". Nature 437 (7057): 376–80. Bibcode 2005Natur.437..376M. doi:10.1038/nature03959. PMC 1464427. PMID 16056220. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1464427.
- ^ Shendure, J.; Porreca, GJ; Reppas, NB; Lin, X; McCutcheon, JP; Rosenbaum, AM; Wang, MD; Zhang, K et al. (2005). "Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome". Science 309 (5741): 1728–32. Bibcode 2005Sci...309.1728S. doi:10.1126/science.1117389. PMID 16081699.
- ^ Applied Biosystems' SOLiD technology
- ^ Braslavsky I, Hebert B, Kartalov E, Quake SR (April 2003). "Sequence information can be obtained from single DNA molecules". Proc. Natl. Acad. Sci. U.S.A. 100 (7): 3960–4. Bibcode 2003PNAS..100.3960B. doi:10.1073/pnas.0230489100. PMC 153030. PMID 12651960. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=153030.
- ^ Hall N (May 2007). "Advanced sequencing technologies and their wider impact in microbiology". J. Exp. Biol. 210 (Pt 9): 1518–25. doi:10.1242/jeb.001370. PMID 17449817.
- ^ Church GM (January 2006). "Genomes for all". Sci. Am. 294 (1): 46–54. doi:10.1038/scientificamerican0106-46. PMID 16468433.
- ^ Schuster, Stephan C. (2008). "Next-generation sequencing transforms today's biology". Nature methods (Nature Methods) 5 (1): 16–18. doi:10.1038/nmeth1156. PMID 18165802.
- ^ Brenner, Sidney; Johnson, M; Bridgham, J; Golda, G; Lloyd, DH; Johnson, D; Luo, S; McCurdy, S et al. (2000). "Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays". Nature Biotechnology (Nature Biotechnology) 18 (6): 630–634. doi:10.1038/76469. PMID 10835600.
- ^ a b c Schuster SC (January 2008). "Next-generation sequencing transforms today's biology". Nat. Methods 5 (1): 16–8. doi:10.1038/nmeth1156. PMID 18165802.
- ^ Mardis ER (2008). "Next-generation DNA sequencing methods". Annu Rev Genomics Hum Genet 9: 387–402. doi:10.1146/annurev.genom.9.081307.164359. PMID 18576944.
- ^ Valouev A, Ichikawa J, Tonthat T, et al. (July 2008). "A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning". Genome Res. 18 (7): 1051–63. doi:10.1101/gr.076463.108. PMC 2493394. PMID 18477713. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2493394.
- ^ Rusk, N. (2011). "Torrents of sequence." Nat Meth 8(1): 44-44.
- ^ Human Genome Sequencing Using Unchained Base Reads in Self-Assembling DNA Nanoarrays. Drmanac, R. et. al. Science, 2010, 327 (5961): 78-81,
- ^ Genome Sequencing on Nanoballs Porreca, JG. Nature Biotechnology, 2010, 28:(43-44)
- ^ Human Genome Sequencing Using Unchained Base Reaads in Self-Assembling DNA Nanoarrays, Supplementary Material. Drmanac, R. et. al. Science, 2010, 327 (5961):78-81,
- ^ Complete Genomics Press release, 2010
- ^ Hanna GJ, Johnson VA, Kuritzkes DR, et al (1 July 2000). "Comparison of Sequencing by Hybridization and Cycle Sequencing for Genotyping of Human Immunodeficiency Virus Type 1 Reverse Transcriptase". J. Clin. Microbiol. 38 (7): 2715–21. PMC 87006. PMID 10878069. http://jcm.asm.org/cgi/pmidlookup?view=long&pmid=10878069.
- ^ J.R. Edwards, H.Ruparel, and J. Ju (2005). "Mass-spectrometry DNA sequencing". Mutation Research 573 (1–2): 3–12. doi:10.1016/j.mrfmmm.2004.07.021. PMID 15829234.
- ^ "VisiGen Biotechnologies Inc. - Technology Overview". Visigenbio.com. http://visigenbio.com/technology_overview.html. Retrieved 2009-11-15.
- ^ "The Harvard Nanopore Group". Mcb.harvard.edu. http://mcb.harvard.edu/branton/index.htm. Retrieved 2009-11-15.
- ^ "Nanopore Sequencing Could Slash DNA Analysis Costs". http://www.physorg.com/news157378086.html.
- ^ US patent 20060029957, ZS Genetics, "Systems and methods of analyzing nucleic acid polymers and related components", issued 2005-07-14
- ^ Xu M, Fujita D, Hanagata N (December 2009). "Perspectives and challenges of emerging single-molecule DNA sequencing technologies". Small 5 (23): 2638–49. doi:10.1002/smll.200900976. PMID 19904762.
- ^ Schadt, E.E.; S. Turner, A. Kasarskis (2010). "A window into third-generation sequencing". Human Molecular Genetics 19 (R2): R227–40. doi:10.1093/hmg/ddq416. PMID 20858600.
- ^ Ying-Ja Chen, Eric E. Roller and Xiaohua Huang (2010). "DNA sequencing by denaturation: experimental proof of concept with an integrated fluidic device". Lab on Chip 10 (10): 1153–1159. doi:10.1039/b921417h.
- ^ "PRIZE Overview: Archon X PRIZE for Genomics"
- ^ The Future of DNA Sequencing
- ^ Watson JD, Crick FH (1953). "The structure of DNA". Cold Spring Harb. Symp. Quant. Biol. 18: 123–31. PMID 13168976.
- ^ Sanger F, Air GM, Barrell BG, et al. (February 1977). "Nucleotide sequence of bacteriophage phi X174 DNA". Nature 265 (5596): 687–95. Bibcode 1977Natur.265..687S. doi:10.1038/265687a0. PMID 870828.
- ^ Adams MD, Kelley JM, Gocayne JD, et al. (June 1991). "Complementary DNA sequencing: expressed sequence tags and human genome project". Science 252 (5013): 1651–6. Bibcode 1991Sci...252.1651A. doi:10.1126/science.2047873. PMID 2047873.
- ^ Fleischmann RD, Adams MD, White O, et al. (July 1995). "Whole-genome random sequencing and assembly of Haemophilus influenzae Rd". Science 269 (5223): 496–512. Bibcode 1995Sci...269..496F. doi:10.1126/science.7542800. PMID 7542800. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=7542800.
- ^ M. Ronaghi, S. Karamohamed, B. Pettersson, M. Uhlen, and P. Nyren (1996). "Real-time DNA sequencing using detection of pyrophosphate release". Analytical Biochemistry 242 (1): 84–9. doi:10.1006/abio.1996.0432. PMID 8923969.
- ^ Ewing B, Green P (March 1998). "Base-calling of automated sequencer traces using phred. II. Error probabilities". Genome Res. 8 (3): 186–94. doi:10.1101/gr.8.3.186 (inactive 2010-01-07). PMID 9521922. http://www.genome.org/cgi/pmidlookup?view=long&pmid=9521922.
- ^ Brenner S, et al. (2000). "Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays". Nature Biotechnology (Nature Biotechnology) 18 (6): 630–634. doi:10.1038/76469. PMID 10835600.
- ^ Lander ES, Linton LM, Birren B, et al. (February 2001). "Initial sequencing and analysis of the human genome". Nature 409 (6822): 860–921. doi:10.1038/35057062. PMID 11237011.
- ^ Venter JC, Adams MD, Myers EW, et al. (February 2001). "The sequence of the human genome". Science 291 (5507): 1304–51. Bibcode 2001Sci...291.1304V. doi:10.1126/science.1058040. PMID 11181995.
- ^ Stein RA (1 September 2008). "Next-Generation Sequencing Update". Genetic Engineering & Biotechnology News 28 (15). http://www.genengnews.com/gen-articles/next-generation-sequencing-update/2584/.
- ^ Margulies M, Egholm M, Altman WE, et al. (September 2005). "Genome Sequencing in Open Microfabricated High Density Picoliter Reactors". Nature 437 (7057): 376–80. Bibcode 2005Natur.437..376M. doi:10.1038/nature03959. PMC 1464427. PMID 16056220. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1464427.
Categories:- Molecular biology
- DNA sequencing
- Molecular biology techniques

Wikimedia Foundation. 2010.