- Cytochrome P450 reductase
-
Cytochrome P450 reductase (EC 1.6.2.4; also known as NADPH:ferrihemoprotein oxidoreductase, NADPH:hemoprotein oxidoreductase, NADPH:P450 oxidoreductase, P450 reductase, POR, CPR, CYPOR) is a membrane-bound enzyme required for electron transfer to cytochrome P450 in the microsome of the eukaryotic cell from a FAD- and FMN-containing enzyme NADPH:cytochrome P450 reductase (POR; EC 1.6.2.4).
Contents
Introduction
In Bacillus megaterium and Bacillus subtilis, POR is a C-terminal domain of CYP102, a single-polypeptide self-sufficient soluble P450 system (P450 is an N-terminal domain). The general scheme of electron flow in the POR/P450 system is:
NADPH → FAD → FMN → P450 → O2 The definitive evidence for the requirement of POR in cytochrome-P450-mediated reactions came from the work of Lu, Junk and Coon,[1] who dissected the P450-containing mixed function oxidase system into three constituent components: POR, cytochrome P450, and lipids.
Since all microsomal P450 enzymes require POR for catalysis, it is expected that disruption of POR would have devastating consequences. POR knockout mice are embryonic lethal,[2] probably due to lack of electron transport to extrahepatic P450 enzymes since liver-specific knockout of POR yields phenotypically and reproductively normal mice that accumulate hepatic lipids and have remarkably diminished capacity of hepatic drug metabolism.[3]
The reduction of cytochrome P450 is not the only physiological function of POR. The final step of heme oxidation by mammalian heme oxygenase requires POR and O2. In yeast, POR affects the ferrireductase activity, probably transferring electrons to the flavocytochrome ferric reductase.[4]
Gene organization
Human POR gene has 16 exons and the exons 2-16 code for a 677-amino acid [5] POR protein (NCBI NP_000932.2). There is a single copy of 50 kb POR gene (NCBI NM_000941.2) in humans on chromosome 7 (7q11.23).
Mutations and polymorphisms
Flück et al.[6] have reported for the first time five missense mutations (A284P, R454H, V489E, C566Y, and V605F) and a splicing mutation in the POR genes of four patients who had hormonal evidence for combined deficiencies of two steroidogenic cytochrome P450 enzymes - P450c17 CYP17A1, which catalyzes steroid 17α-hydroxylation and 17,20 lyase reaction, and P450c21 21-Hydroxylase, which catalyzes steroid 21-hydroxylation. Later on Arlt et al.[7] identified another POR missense mutation, Y178D, and also reported three of the POR mutations (A284P, R454H, and C566Y), that were originally described by Flück et al.. In a larger study Huang et al.[8] have examined the POR genes in 32 additional patients. Fifteen of nineteen patients having abnormal genitalia and disordered steroidogenesis were homozygous or apparent compound heterozygous for POR mutations that destroyed or dramatically inhibited POR activity. Huang et al.. studied 11 new POR variants: A115V, T142A, Q153R, P228L, M263V, R316W, G413S, Y459H, A503V, G504R, G539R, L565P, R616X, V631I, and F646del.
POR Deficiency – Mixed Oxidase Disease
POR deficiency is the newest form of congenital adrenal hyperplasia first described in 2004.[6] The index patient was a newborn 46,XX Japanese girl with craniosynostosis, hypertelorism, mid-face hypoplasia, radiohumeral synostosis, arachnodactyly and disordered steroidogenesis. However, the clinical and biochemical characteristics of patients with POR deficiency are long known in the literature as so-called mixed oxidase disease, as POR deficiency typically shows a steroid profile that suggests combined deficiencies of steroid 21-hydroxylase and 17α-hydroxylase/17,20 lyase activities. The clinical spectrum of POR deficiency ranges from severely affected children with ambiguous genitalia, adrenal insufficiency, and the Antley-Bixler skeletal malformation syndrome (ABS) to mildly affected individuals with polycystic ovary syndrome-like features. Some of the POR patients were born to mothers who became virilized during pregnancy, suggesting deficient placental aromatization of fetal androgens due to a lesion in microsomal aromatase resulting in low estrogen production, which was later confirmed by lower aromatase activities caused by POR mutations.[9] However, it has also been suggested that fetal and maternal virilization in POR deficiency might be caused by increased dihydrotestosterone synthesis by the fetal gonad through an alternative "backdoor" pathway first described in the marsupials. Gas chromatography/mass spectrometry analysis of urinary steroids from pregnant women carrying a POR-deficient fetus supports the existence of this pathway,[10] but the relevance of the "backdoor" pathway in the normal or CAH fetus remains unclear. The role of POR mutations beyond CAH remains unknown; and questions such as how POR mutations cause bony abnormalities and what role POR variants play in drug metabolism by hepatic P450s are unsolved. However, reports of ABS in some offsprings of mothers who were treated with fluconazole, an antifungal agent which interferes with cholesterol biosynthesis at the level of CYP51 activity - indicate that disordered drug metabolism may result from deficient POR activity.
Williams Syndrome
Williams syndrome is a genetic disorder characterized by the deletion of genetic material approximately 1.2 Mb from the POR gene (POR). Cells with this genetic deletion show reduced transcription of POR, it seems, due to the loss of a cis-regulatory element that alters expression of this gene.[11] Some persons with Williams syndrome show characteristics of POR deficiency, including radio-ulnar synostosis and other skeletal abnormalities.[12] Cases of mild impairment of cortisol and androgen synthesis have been noted,[13] however, despite the fact that deficient POR impairs androgen synthesis, patients with Williams syndrome often show increased androgen levels.[14] A similar increase in testosterone has been observed in a mouse model that has globally decreased POR expression.[15]
Structure
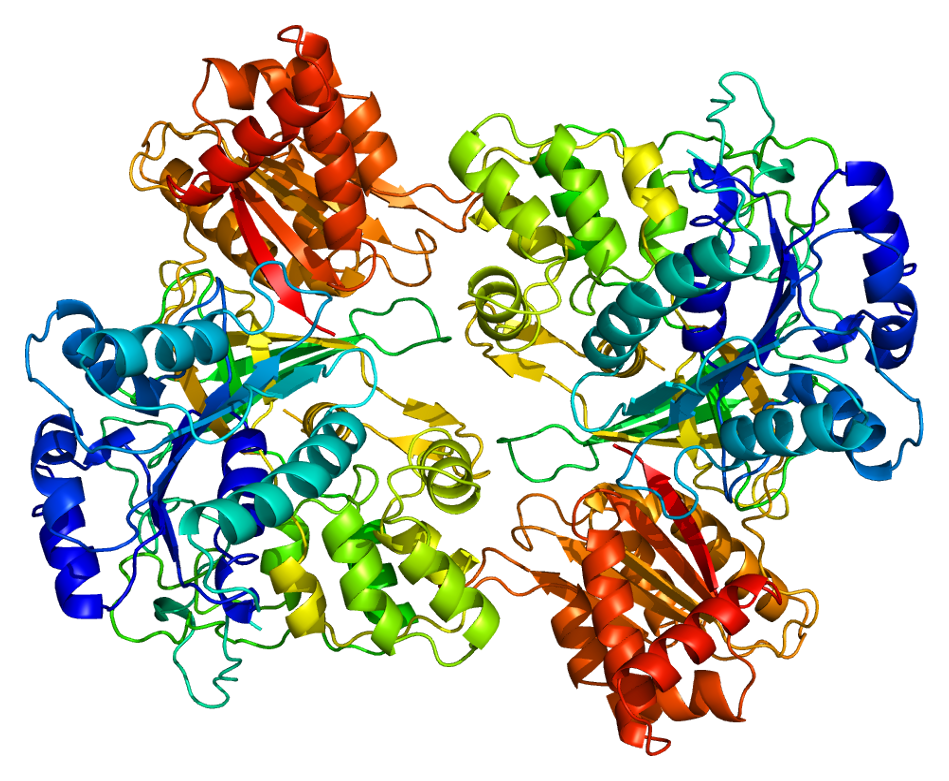
3D crystal structure of rat POR has been solved[16] (PDB 1AMO). The molecule is composed of four structural domains: the FMN-binding domain, the connecting domain, the FAD-binding domain, and NADPH-binding domain. The FMN-binding domain is similar to the structure of FMN-containing protein flavodoxin, whereas the FAD-binding domain and NADPH-binding domains are similar to those of flavoprotein ferredoxin-NADP+ reductase (FNR). The connecting domain is situated between the flavodoxin-like and FNR-like domains.
POR homologs
The other enzymes containing homologs of POR are nitric oxide synthase (EC 1.14.13.39), NADPH:sulfite reductase (EC 1.8.1.2), and methionine synthase reductase (EC 1.16.1.8).
See also
References
- ^ Lu, A.Y.H., Junk, K.W. and Coon, M.J. (1969). "Resolution of the cytochrome P-450-containing ω-hydroxylation system of liver microsomes into three components". J. Biol. Chem. 244 (13): 3714–3721. PMID 4389465.
- ^ Shen, A.L., O'Leary, K.A. and Kasper, C.B. (2002). "Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase". J. Biol. Chem. 277 (8): 6536–6541. doi:10.1074/jbc.M111408200. PMID 11742006.
- ^ Gu, J., Weng, Y., Zhang, Q.-Y., Cui, H., Behr, M., Wu, L., Yang, W., Zhang, L. and Ding, X. (2003). "Liver-specific deletion of the NADPH-cytochrome P450 reductase gene. Impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase". J. Biol. Chem. 278 (28): 25895–25901. doi:10.1074/jbc.M303125200. PMID 12697746.
- ^ Lesuisse, E., Casteras-Simon, M. and Labbe, P. (1997). "Cytochrome P-450 reductase is responsible for the ferrireductase activity associated with isolated plasma membranes of Saccharomyces cerevisiae". FEMS Microbiol. Lett. 156 (1): 147–152. doi:10.1016/S0378-1097(97)00418-7. PMID 9368374.
- ^ Haniu, M., McManus, M.E., Birkett, D.J., Lee, T.D. and Shively, J.E. (1989). "Structural and functional analysis of NADPH-cytochrome P-450 reductase from human liver: complete sequence of human enzyme and NADPH-binding sites". Biochemistry 28 (21): 8639–8645. doi:10.1021/bi00447a054. PMID 2513880.
- ^ a b Flück, C.E., Tajima, T., Pandey, A.V., Arlt, W., Okuhara, K., Verge, C.F., Jabs, E.W., Mendonça, B.B., Fujieda, K. and Miller, W.L. (2004). "Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome". Nature Genetics 36 (3): 228–230. doi:10.1038/ng1300. PMID 14758361.
- ^ Arlt, W., Walker, E.A., Draper, N., Ivison, H.E., Ride, J.P., Hammer, F., Chalder, S.M., Borucka-Mankiewicz, M., Hauffa, B.P., Malunowicz, E.M., Stewart, P.M. and Shackleton, C.H.L. (2004). "Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study". Lancet 363 (9427): 2128–2135. doi:10.1016/S0140-6736(04)16503-3. PMID 15220035.
- ^ Huang, N., Pandey, A.V., Agrawal, V., Reardon, W., Lapunzina, P.D., Mowat, D., Jabs, E.W., Van Vliet, G., Sack, J., Flück, C.E. and Miller, W.L. (2005). "Diversity and function of mutations in P450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis". Am. J. Hum. Genet. 76 (5): 729–749. doi:10.1086/429417. PMC 1199364. PMID 15793702. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1199364.
- ^ Pandey AV, Kempná P, Hofer G, Mullis PE, Flück CE (October 2007). "Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase". Mol. Endocrinol. 21 (10): 2579–95. doi:10.1210/me.2007-0245. PMID 17595315. http://www.ncbi.nlm.nih.gov/pubmed/1759531.
- ^ Shackleton, C., Marcos, J., Arlt, W. and Hauffa, B.P. (2004). "Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): a disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype". Am. J. Med. Genet. A 129 (2): 105–112. doi:10.1002/ajmg.a.30171. PMID 15316970.
- ^ Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, Zabot MT, Antonarakis SE, Reymond A (2006). "Submicroscopic Deletion in Patients with Williams-Beuren Syndrome Influences Expression Levels of the Nonhemizygous Flanking Genes". Am J Hum Genet 79 (2): 332–341. doi:10.1086/506371. PMC 1559497. PMID 16826523. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1559497.
- ^ Charvat KA, Hornstein L, Oestreich AE (1991). "Radio-ulnar synostosis in Williams syndrome. A frequently associated anomaly.". Pediatr Radiol 21 (7): 508–10. PMID 1771116.
- ^ Ichinose M, Tojo K, Nakamura K, Matsuda H, Tokudome G, Ohta M, Sakai S, Sakai O (1996). "Williams syndrome associated with chronic renal failure and various endocrinological abnormalities". Intern Med 35 (6): 482–8. doi:10.2169/internalmedicine.35.482. PMID 8835601.
- ^ Partsch CJ, Pankau R, Blum WF, Gosch A, Wessel A (1994). "Hormonal regulation in children and adults with Williams-Beuren syndrome". Am J Med Genet 51 (3): 251–7. doi:10.1002/ajmg.1320510316. PMID 8074154.
- ^ Wu L, Gu J, Cui H, Zhang QY, Behr M, Fang C, Weng Y, Kluetzman K, Swiatek PJ, Yang W, Kaminsky L, Ding X (2005). "Transgenic Mice with a Hypomorphic NADPH-Cytochrome P450 Reductase Gene: Effects on Development, Reproduction, and Microsomal Cytochrome P450". J Pharmacol Exp Ther. 312 (1): 35–43. doi:10.1124/jpet.104.073353. PMID 15328377.
- ^ Wang, M., Roberts, D.L., Paschke, R., Shea, T.M., Masters, B.S.S. and Kim, J.-J.P. (1997). "Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes". Proc. Natl. Acad. Sci. USA 94 (16): 8411–8416. doi:10.1073/pnas.94.16.8411. PMC 22938. PMID 9237990. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=22938.
External links
- MeSH Cytochrome+P450+Reductase
- GeneReviews/NCBI/NIH/UW entry on Cytochrome P450 Oxidoreductase Deficiency
Human physiology: Endocrinology Fields Other Blood sugar regulation • Calcium metabolism • Endocrine glands • Wolff–Chaikoff effect/Jod-Basedow effectAcetolactate synthase - Acyl CoA dehydrogenase - Apoptosis-inducing factor - Butyryl CoA dehydrogenase - Cryptochrome - Cytochrome b5 reductase - Dihydrolipoamide dehydrogenase - Flavodoxin - Methemoglobin reductase - Methylenetetrahydrofolate reductase - NADH dehydrogenase - NADPH oxidase - Nitrate reductase - Sarcosine oxidase - Thioredoxin reductaseOxidoreductases: NADH or NADPH (EC 1.6) 1.6.1: NAD/NADP 1.6.2: Heme Methemoglobin reductase · NADPH-hemoprotein reductase/Cytochrome P450 reductase · NADPH-cytochrome-c2 reductase · Leghemoglobin reductase1.6.3: Oxygen 1.6.5: Quinone or similar 1.6.6: Nitrogenous group 1.6.99: other Categories:- Human proteins
- Cytochrome P450

Wikimedia Foundation. 2010.