- Malpuech facial clefting syndrome
-
Malpuech facial clefting syndrome Classification and external resources ICD-10 Q89.8 ICD-9 759.7 OMIM 248340 Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome,[1] is a rare congenital syndrome. It is characterized by facial clefting (any type of cleft in the bones and tissues of the face, including a cleft lip and palate), a caudal appendage (a "human tail"),[2][3] growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia.[4] Abnormalities of the heart, and other skeletal malformations may also be present.[5] The syndrome was initially described by Guilliaume Malpuech and associates in 1983.[6] It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes.[7][8] Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes.[9] The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.[10]
Contents
Characteristics
 A typical cleft lip, seen in a five-month-old infant.
A typical cleft lip, seen in a five-month-old infant. The bilateral type of cleft lip has been reported in Malpuech syndrome.
The bilateral type of cleft lip has been reported in Malpuech syndrome.Malpuech syndrome is congenital, being apparent at birth. It is characterized by a feature known as facial clefting. Observed and noted in the initial description of the syndrome as a cleft lip and palate,[6] facial clefting is identified by clefts in the bones, muscles and tissues of the face, including the lips and palate. The forms of cleft lip and palate typically seen with Malpuech syndrome are midline (down the middle of the lip and palate)[11] or bilateral (affecting both sides of the mouth and palate).[10] Facial clefting generally encompasses a wide range of severity, ranging from minor anomalies such as a bifid (split) uvula, to a cleft lip and palate, to major developmental and structural defects of the facial bones and soft tissues.[12] Clefting of the lip and palate occurs during embryogenesis.[13][14] Additional facial and ortho-dental anomalies that have been described with the syndrome include: hypertelorism (unusually wide-set eyes, sometimes reported as telecanthus), narrow palpebral fissures (the separation between the upper and lower eyelids) and ptosis (drooping) of the eyelids, frontal bossing (prominent eyebrow ridge) with synophris, highly arched eyebrows, wide nasal root and a flattened nasal tip, malar hypoplasia (underdeveloped upper cheek bone), micrognathia (an undersized lower jaw), and prominent incisors. Auditory anomalies include an enlarged ear ridge, and hearing impairment associated with congenital otitis media (or "glue ear", inflammation of the middle ear) and sensorineural hearing loss.[4][15][16]
Another feature identified with Malpuech syndrome is a caudal appendage.[1][3] A caudal appendage is a congenital outgrowth stemming from the coccyx (tailbone). Present in many non-human animal species as a typical tail, this feature when seen in an infant has been described as a "human tail".[2][17] This was observed by Guion-Almeida (1995) in three individuals from Brazil. The appendage on X-rays variously appeared as a prominent protrusion of the coccyx.[3] On a physical examination, the appendage resembles a nodule-like stub of an animal tail.[18]
Deficiencies such as mental retardation, learning disability, growth retardation and developmental delay are common. Psychiatric manifestations that have been reported with the syndrome include psychotic behavior, obsessive–compulsive disorder, loss of inhibition, hyperactivity, aggression, fear of physical contact, and compulsive actions like echolalia (repeating the words spoken by another person). Neuromuscular tics have also been noted.[4][15]
Urogenital abnormalities, or those affecting the urinary and reproductive systems, are common with the syndrome. Malpuech et al. (1983) and Kerstjens-Frederikse et al. (2005) reported variously in affected males a micropenis, hypospadias (a congenital mislocation of the urinary meatus), cryptorchidism (ectopic or undescended testes), bifid (split) and underdeveloped scrotum, and an obstructive urethral valve.[4][6] An affected boy was also reported by Reardon et al. (2001) with left renal agenesis, an enlarged and downwardly displaced right kidney, cryptorchidism and a shawl scrotum.[7] Other malformations that have been noted with the syndrome are omphalocele[3] and an umbilical hernia.[19]
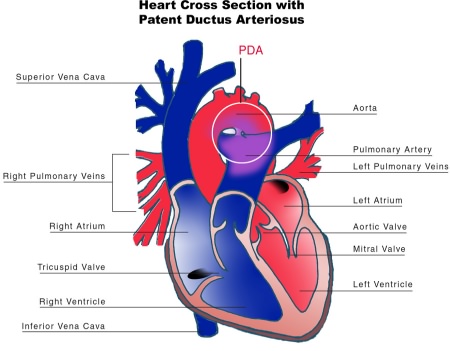
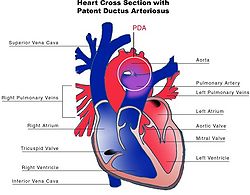 Diagram of a neonatal heart affected by PDA
Diagram of a neonatal heart affected by PDA
Congenital abnormalities of the heart have also been observed with Malpuech syndrome. From a healthy Japanese couple, Chinen and Naritomi (1995) described the sixth child who had features consistent with the disorder. This two-month-old male infant was also affected by cardiac anomalies including patent ductus arteriosus (PDA)[20] and ventricular septal defect.[5] The opening in the ductus arteriosus associated with PDA had been surgically repaired in the infant at 38 days of age. A number of minor skeletal aberrations were also reported in the infant, including wormian bones at the lambdoid sutures.[5]
Classification
Malpuech syndrome has been shown to have physical, or phenotypical similarities with several other genetic disorders. A report by Reardon et al. (2001) of a nine-year-old boy exhibiting facial, caudal and urogenital anomalies consistent with Malpuech syndrome, who also had skeletal malformites indicative of Juberg-Hayward syndrome, suggests that the two disorders may be allelic (caused by different mutations of the same gene).[7]
Along with several other disorders that have similar, or overlapping features and autosomal recessive inheritance, Malpuech syndrome has been considered to belong under the designation "3MC syndrome". Titomanlio et al. (2005) described a three-year-old female known to have Michels syndrome. In their review of the physical similarities between Michels, Malpuech and Mingarelli-Carnevale syndromes—particularly the facial appearance including instances of cleft lip and palate, and ptosis, and a similarity of congenital abdominal and urogenital anomalies—they believed the syndromes may represent a spectrum of genetic disorders rather than three individual disorders. They initially suggested this spectrum could be named 3MC (Michels-Malpuech-Mingarelli-Carnevale) syndrome.[8] This conclusion and the name 3MC syndrome was supported by Leal et al. (2008), who reported a brother and sister with an array of symptoms that overlapped the various syndromes.[21] Further assertion of 3MC syndrome was by Rooryck et al. (2011) in an elaboration of its cause.[9]
Cause and genetics
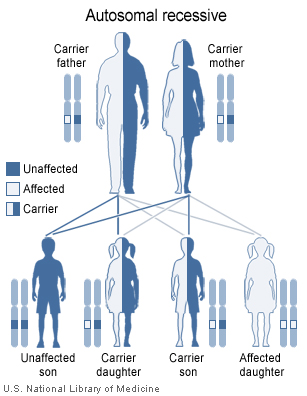
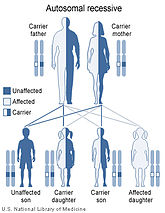 Malpuech facial clefting syndrome has an autosomal recessive pattern of inheritance.
Malpuech facial clefting syndrome has an autosomal recessive pattern of inheritance.Malpuech syndrome, as with the other disorders within the 3MC syndrome consideration, is caused by mutations in the COLLEC11 and MASP1 genes. In an investigation by Rooryck et al. (2011), eleven families affected by 3MC syndrome were studied, which resulted in the identification of these two mutations. Both genes encode proteins of the lectin complement pathway, which plays a role in the complement system of innate, or non-specific immunity in humans and other species.[9]
The COLLEC11, or CL-K1 gene is located on the short arm of chromosome 2 (2p25.3) in humans.[22] The CL-K1 protein is a C-type lectin, and belongs to the collectin family of these proteins. Other than its role in innate immunity, the protein is thought to be involved in the development of tissues including craniofacial cartilage, the heart and kidney during embryogenesis. This function in facial development was corroborated through study of the zebrafish, where mutations in its version of CL-K1 contributed to craniofacial abnormalities possibly associated with errors in neural crest cell migration.[9]
 The short and long arm of a typical human chromosome.
The short and long arm of a typical human chromosome.The MASP1, or Mannan-binding Serine Protease I gene is located on the long arm of human chromosome 3 at 3q27-q28.[23] The protein is a type of connectin called a mannan-binding lectin, which plays a role in innate immunity by binding to pathogens such as viruses including HIV.[24]
As described by Sirmaci et al. (2010), three Turkish individuals from two consanguineous families (the children of relatives such as cousins are said to be in a consanguineous family) with various characteristics of 3MC syndrome, including facial dysmorphism and a caudal appendage, were evaluated. Investigation of homologous chromosomes through gene mapping revealed an autozygous region (a location on a chromosome where both alleles of a gene originate from a common ancestor) at chromosome 3q27 in both families. In one family, a missense mutation in MASP1 at this location resulted in the replacement of the amino acid glycine by arginine at position 687 in the gene sequence. The mutation cosegregated with the observed phenotype. In individuals from the second family, DNA sequencing of MASP1 showed a nonsense mutation that resulted in a deactivation of tryptophan at position 290 in the gene, that also cosegregated with the phenotype. Both mutations occur in a form of MASP1 known to process IGFBP5; loss of this function associated with mutation of MASP1 causes disruptions in the availability of insulin-like growth factor during craniofacial and musculoskeletal development during the embryonic period. These results indicate that mutations in MASP1 are responsible for an array of features found with malformation disorders including Malpuech syndrome.[25]
The syndrome is inherited in an autosomal recessive manner.[10] This means the defective gene(s) responsible for the disorder (COLLEC11, MASP1) is located on an autosome (chromosomes 2 and 3 are autosomes), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Diagnosis
It is suggested that the diagnostic criteria for Malpuech syndrome should include cleft lip and/or palate, typical associated facial features, and at least two of the following: urogenital anomalies, caudal appendage, and growth or developmental delay.[26] Due to the relatively high rate of hearing impairment found with the disorder, it too may be considered in the diagnosis. Another congenital disorder, Wolf-Hirschhorn (Pitt-Rogers-Danks) syndrome, shares Malpuech features in its diagnostic criteria. Because of this lacking differentiation, karyotyping (microscopic analysis of the chromosomes of an individual) can be employed to distinguish the two. Whereas deletions in the short arm of chromosome 4 would be revealed with Wolf-Hirschhorn, a karyotype without this aberration present would favor a Malpuech syndrome diagnosis. Also, the karyotype of an individual with Malpuech syndrome alone will be normal.[15]
Management
Many of the congenital malformations found with Malpuech syndrome can be corrected surgically. These include cleft lip and palate, omphalocele, urogenital and craniofacial abnormalities, skeletal deformities such as a caudal appendage or scoliosis, and hernias of the umbillicus. The primary area of concern for these procedures applied to a neonate with congenital disorders including Malpuech syndrome regards the logistics of anesthesia. Methods like tracheal intubation for management of the airway during general anesthesia can be hampered by the even smaller, or maldeveloped mouth of the infant. For regional anesthesia, methods like spinal blocking are more difficult where scoliosis is present. In a 2010 report by Kiernan et al., a four-year-old girl with Malpuech syndrome was being prepared for an unrelated tonsillectomy and adenoidectomy. While undergoing intubation, insertion of a laryngoscope, needed to identify the airway for the placement of the endotracheal tube, was made troublesome by the presence of micrognathia attributed to the syndrome. After replacement with a laryngoscope of adjusted size, intubation proceeded normally. Successful general anesthesia followed.[16]
A rare follow-up of a male with Malpuech syndrome was presented by Priolo et al. (2007). Born at term from an uneventful pregnancy and delivery, the infant underwent a surgical repair of a cleft lip and palate. No problems were reported with the procedure. A heart abnormality, atrial septal defect, was also apparent but required no intervention. At age three years, mental retardation, hyperactivity and obsessive compulsive disorder were diagnosed; hearing impairment was diagnosed at age six, managed with the use of hearing aids. Over the course of the decade that followed, a number of psychiatric evaluations were performed. At age 14, he exhibited a fear of physical contact; at age 15, he experienced a severe psychotic episode, characterized by agitation and a loss of sociosexual inhibition. This array of symptoms were treated pharmocologically (with prescription medications). He maintained a low level of mental deficiency by age 17, with moments of compulsive echolalia.[15]
Epidemiology and history
The incidence of Malpuech syndrome has not been determined.[16] A 1999 report by Crisponi et al. suggested that only about 12 individuals worldwide were affected by the disorder at that time.[19] The syndrome was first reported by Guilliaume Malpuech and colleagues in 1983, observed in four children of unspecified gender in what was described as a gypsy family. The children included three siblings and their first cousin; the family was known to be highly consanguineous.[6][15]
References
- ^ a b Online 'Mendelian Inheritance in Man' (OMIM) Malpuech facial clefting syndrome -248340
- ^ a b Adeleye, A. O.; Olowookere, K. G. (Sep-Oct 2010). "A Human Tail in an Infant (Letter)". West African Journal of Medicine 29 (5): 356–358. PMID 21089026.
- ^ a b c d Guion‐Almeida, M. L. (Jul 1995). "Apparent Malpuech syndrome: Report on three Brazilian patients with additional signs". American Journal of Medical Genetics 58 (1): 13–17. doi:10.1002/ajmg.1320580104. PMID 7573149.
- ^ a b c d Kerstjens-Frederikse, W. S.; Brunner, H. G.; Van Dael, C. M.; Van Essen, A. J. (May 2005). "Malpuech syndrome: Three patients and a review". American Journal of Medical Genetics Part A 134 (4): 450–453. doi:10.1002/ajmg.a.30662. PMID 15793834.
- ^ a b c Chinen, Y.; Naritomi, K. (Dec 1995). "Malpuech facial clefting syndrome in a Japanese boy with cardiac defects". The Japanese Journal of Human Genetics 40 (4): 335–338. doi:10.1007/BF01900601. PMID 8851768.
- ^ a b c d Malpuech, G.; Demeocq, F.; Palcoux, J. B.; Vanlieferinghen, P.; Opitz, J. M. (Dec 1983). "A previously undescribed autosomal recessive multiple congenital anomalies/mental retardation (MCA/MR) syndrome with growth failure, lip/palate cleft(s), and urogenital anomalies". American Journal of Medical Genetics 16 (4): 475–480. doi:10.1002/ajmg.1320160405. PMID 6660246.
- ^ a b c Reardon, W.; Hall, C. M.; Gorman, W. (Apr 2001). "An atypical case suggesting the possibility of overlap between Malpuech and Juberg-Hayward syndromes". Clinical dysmorphology 10 (2): 123–128. doi:10.1097/00019605-200104000-00009. PMID 11310992.
- ^ a b Titomanlio, L.; Bennaceur, S.; Bremond-Gignac, D.; Baumann, C.; Dupuy, O.; Verloes, A. (Sep 2005). "Michels syndrome, Carnevale syndrome, OSA syndrome, and Malpuech syndrome: Variable expression of a single disorder (3MC syndrome)?". American Journal of Medical Genetics Part A 137A (3): 332–335. doi:10.1002/ajmg.a.30878. PMID 16096999.
- ^ a b c d Rooryck, C.; Diaz-Font, A.; Osborn, D. P.; Chabchoub, E.; Hernandez-Hernandez, V.; Shamseldin, H.; Kenny, J.; Waters, A. et al. (Mar 2011). "Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome". Nature Genetics 43 (3): 197–203. doi:10.1038/ng.757. PMID 21258343.
- ^ a b c Turnbull, C.; Lees, M.; Chitty, L. S. (Dec 2006). "Prenatal sonographic diagnosis of Malpuech syndrome". Prenatal Diagnosis 26 (12): 1121–1123. doi:10.1002/pd.1564. PMID 17019743.
- ^ "Midline cleft lip in children". Wrongdiagnoysis.com. http://www.wrongdiagnosis.com/sym/midline_cleft_lip_in_children.htm. Retrieved February 27, 2011.
- ^ "FACE - DIAGNOSIS OF CONGENITAL ABNORMALITIES - THE 18-23 WEEKS SCAN". Centrus.com.br. http://www.centrus.com.br/DiplomaFMF/SeriesFMF/18-23-weeks/chapter-03/face/facefmf.html. Retrieved November 16, 2010.
- ^ "Definition of facial clefting". Medilexicon.com. http://www.medilexicon.com/medicaldictionary.php?t=18132. Retrieved November 7, 2010.
- ^ "Rare facial cleft: 14-mth-old Hunan boy Kang Kang born with a 'mask'". Whatsonxiamen.com. http://www.whatsonxiamen.com/news12345.html. Retrieved November 8, 2010.
- ^ a b c d e Priolo, M; Ciccone, R; Bova, I; Campolo, G; Lagana, C; Zuffardi, O (Mar-Apr 2007). "Malpuech syndrome: Broadening the clinical spectrum and molecular analysis by array-CGH". European Journal of Medical Genetics 50 (2): 139–143. doi:10.1016/j.ejmg.2006.10.004. PMID 17140870.
- ^ a b c Kiernan, F; Crowe, S (Apr 2010). "Malpuech syndrome: implications for anesthetic management". Pediatric Anesthesia 20 (4): 370–371. doi:10.1111/j.1460-9592.2010.03271.x. PMID 20470345.
- ^ Singh, D.; Kumar, B.; Sinha, V.; Bagaria, H. (Sep 2008). "The human tail: rare lesion with occult spinal dysraphism—a case report". Journal of Pediatric Surgery 43 (9): e41–43. doi:10.1016/j.jpedsurg.2008.04.030. PMID 18778987.
- ^ Finn and Lynch (2006), illustration, p. 243.
- ^ a b Crisponi, G; Marras, AR; Corrias, A (Sep 1999). "Two sibs with Malpuech syndrome". American Journal of Medical Genetics 86 (3): 294–299. doi:10.1002/(SICI)1096-8628(19990917)86:3<294::AID-AJMG20>3.0.CO;2-2. PMID 10482884.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) Patent ductus arteriosus -607411
- ^ Leal, G. F.; Silva, E. O.; Duarte, A. R.; Campos, J. F. (Apr 2008). "Blepharophimosis, blepharoptosis, defects of the anterior chamber of the eye, caudal appendage, radioulnar synostosis, hearing loss and umbilical anomalies in sibs: 3MC syndrome?". American Journal of Medical Genetics Part A 146A (8): 1059–1062. doi:10.1002/ajmg.a.32252. PMID 18266249.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) COLLEC11 -612502
- ^ Online 'Mendelian Inheritance in Man' (OMIM) MASP1 -600521
- ^ Ji, X; Gewurz, H; Spear, G (2005). "Mannose binding lectin (MBL) and HIV". Molecular Immunology 42 (2): 145. doi:10.1016/j.molimm.2004.06.015. PMID 15488604.
- ^ Sirmaci, Asli; Walsh, Tom; Akay, Hatice; Spiliopoulos, Michail; Şakalar, Yıldırım Bayezit; Hasanefendioğlu-Bayrak, Aylin; Duman, Duygu; Farooq, Amjad et al. (2010). "MASP1 Mutations in Patients with Facial, Umbilical, Coccygeal, and Auditory Findings of Carnevale, Malpuech, OSA, and Michels Syndromes". The American Journal of Human Genetics 87 (5): 679–86. doi:10.1016/j.ajhg.2010.09.018. PMC 2978960. PMID 21035106. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2978960.
- ^ Finn, S. M.; Lynch, S. A. (Oct 2006). "Malpuech syndrome: facial features in the absence of clefting". Clinical Dysmorphology 15 (4): 243–244. doi:10.1097/01.mcd.0000220621.85896.46. PMID 16957483.
Categories:- Autosomal recessive disorders
- Rare diseases
- Congenital disorders
- Syndromes

Wikimedia Foundation. 2010.