- Holoprosencephaly
-
Holoprosencephaly Classification and external resources 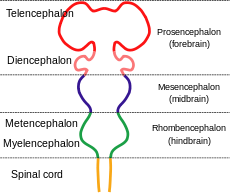
Diagram depicting the main subdivisions of the embryonic vertebrate brain.ICD-10 Q04.2 ICD-9 742.2 OMIM 236100 DiseasesDB 29610 eMedicine radio/347 MeSH D016142 Holoprosencephaly (HPE, once known as arhinencephaly) is a cephalic disorder in which the prosencephalon (the forebrain of the embryo) fails to develop into two hemispheres. Normally, the forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. Hox genes, which guide placement of embryonic structures, fail to activate along the midline of the head, allowing structures that are normally paired on the left and right to merge. The condition also occurs in other species, as with Cy, the Cyclops kitten.
The condition can be mild or severe. According to the National Institute of Neurological Disorders and Stroke (NINDS), "in most cases of holoprosencephaly, the malformations are so severe that babies die before birth.[1]
When the embryo's forebrain does not divide to form bilateral cerebral hemispheres (the left and right halves of the brain), it causes defects in the development of the face and in brain structure and function.
In less severe cases, babies are born with normal or near-normal brain development and facial deformities that may affect the eyes, nose, and upper lip.
Contents
Symptoms
Symptoms of holoprosencephaly range from mild (no facial/organ defects, anosmia, or only a single central incisor) to moderate (cleft lip or cleft palate) to severe (being a cyclops).
There are four classifications of holoprosencephaly.
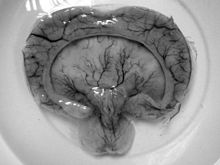 Gross pathology specimen from a case of alobar holoprosencephaly.
Gross pathology specimen from a case of alobar holoprosencephaly.
- Alobar holoprosencephaly, the most serious form, in which the brain fails to separate, is usually associated with severe facial anomalies, including lack of a nose and the eyes merged to a single median structure, see Cyclopia
- Semilobar holoprosencephaly, in which the brain's hemispheres have a slight tendency to separate, is an intermediate form of the disease.
- Lobar holoprosencephaly, in which there is considerable evidence of separate brain hemispheres, is the least severe form. In some cases of lobar holoprosencephaly, the patient's brain may be nearly normal.
- Syntelencephaly, or middle interhemispheric variant of holoprosencephaly (MIHV), in which the posterior frontal lobe and the parietal lobe are not properly separated, but the rostrobasal forebrain properly separates; it is possible that this is not a variant of HPE at all, but is currently classified as such[2]
Presentation
Holoprosencephaly consists of a spectrum of defects or malformations of the brain and face. At the most severe end of this spectrum are cases involving serious malformations of the brain, malformations so severe that they often cause miscarriage or stillbirth. At the other end of the spectrum are individuals with facial defects which may affect the eyes, nose, and upper lip - and normal or near-normal brain development. Seizures and mental retardation may occur.
The most severe of the facial defects (or anomalies) is cyclopia, an abnormality characterized by the development of a single eye, located in the area normally occupied by the root of the nose, and a missing nose or a nose in the form of a proboscis (a tubular appendage) located above the eye. The condition is also referred to as cyclocephaly or synophthalmia, and is very rare.
Causes
The exact cause(s) of HPE are yet to be determined, although the presence of toxins may be suspected. However, it often seems that there is no specific cause at all.[3]
Genetics
Armand Marie Leroi describes the cause of cyclopia as a genetic malfunctioning during the process by which the embryonic brain is divided into two.[4] Only later does the visual cortex take recognizable form, and at this point an individual with a single forebrain region will be likely to have a single, possibly rather large, eye (at such a time, individuals with separate cerebral hemispheres would form two eyes)
Increases in expression of such genes as Pax-2, as well as inhibition of Pax-6, from the notochord have been implicated in normal differentiation of cephalic midline structures. Inappropriate expression of any of these genes may result in mild to severe forms of holoprosencephaly.[citation needed] Other candidate genes have been located, including the SHH (holoprosencephaly type 3 aka HPE3), TGIF, ZIC2 and SIX3 genes.[5]
Although many children with holoprosencephaly have normal chromosomes, specific chromosomal abnormalities have been identified in some patients (trisomy of chromosome 13, also known as Patau syndrome). There is evidence that in some families, HPE is inherited (autosomal dominant as well as autosomal or X-linked recessive inheritance)[citation needed]. Features consistent with familial transmission of the disease (e.g., a single central maxillary incisor) should be carefully assessed in parents and family members.
Non-genetic factors
Numerous possible risk factors have been identified, including gestational diabetes, transplacental infections (the "TORCH complex"), first trimester bleeding, and a history of miscarriage.[3][6] As well, the disorder is found twice as often in female babies.[6] However, there appears to be no correlation between HPE and maternal age.[6]
There is evidence of a correlation between HPE and the use of various drugs classified as being potentially unsafe for pregnant and lactating mothers. These include insulin, birth control pills, aspirin, lithium, thorazine, retinoic acid, and the anticonvulsants.[6] There is also a correlation between alcohol consumption and HPE, along with nicotine, the toxins in cigarettes and toxins in cigarette smoke when used during pregnancy).[6]
Prognosis
HPE is not a condition in which the brain deteriorates over time. Although serious seizure disorders, autonomic dysfunction, complicated endocrine disorders and other life-threatening conditions may sometimes be associated with HPE, the mere presence of HPE does not mean that these serious problems will occur or develop over time without any previous indication or warning. These abnormalities are usually recognized shortly after birth or early in life and only occur if areas of the brain controlling those functions are fused, malformed or absent.
Prognosis is dependent upon the degree of fusion and malformation of the brain, as well as other health complications that may be present.
The more severe forms of holoprosencephaly are usually fatal. This disorder consists of a spectrum of defects, malformations and associated abnormalities. Disability is based upon the degree in which the brain is affected. Moderate to severe defects may cause mental retardation, spastic quadriparesis, athetoid movements, endocrine disorders, epilepsy and other serious conditions. Whereas, mild brain defects may only cause learning or behavior problems with few motor impairments.
Seizures may develop over time with the highest risk before 2 years of age and the onset of puberty. Most are managed with one medication or a combination of medications. Typically, seizures that are difficult to control appear soon after birth, requiring more aggressive medication combinations/doses.
Most children with HPE are at risk of having elevated blood sodium levels during moderate-severe illnesses, that alter fluid intake/output, even if they have no previous diagnosis of diabetes insipidus or hypernatremia.
See also
References
- ^ NINDS Holoencephalopathy Information Page
- ^ Totori-Donati, Paolo; Rossi, Andrea; Biancheri, Roberta (2005). "Brain Malformations". In Totori-Donati, Paolo; Rossi, Andrea; Raybaud, C.. Pediatric Neuroradiology: Brain, Head, Neck and Spine. 1. Springer. pp. 92–95. ISBN 3540410775.
- ^ a b The Carter Centers for Brain Research in Holoprosencephaly and Related Malformations. "About Holoprosencephaly". http://www.stanford.edu/group/hpe/about/.
- ^ Armand Marie Leroi, Mutants: On the Form, Varieties and Errors of the Human Body, 2003, Harper Perennial, London. ISBN 0-00-653164-4
- ^ The Carter Center for Research in holoprosencephaly [1] and [2]
- ^ a b c d e Croen, LA; Croen, Lisa A.; Shaw, Gary M.; Lammer, Edward J. (2000). "Risk Factors For Cytogenetically Normal Holoprosencephaly in California: A Population-Based Case-Control Study". American Journal of Medical Genetics (Wiley-Liss) 90 (4): 320–325. doi:10.1002/(SICI)1096-8628(20000214)90:4<320::AID-AJMG11>3.0.CO;2-8. PMID 10710231.
External links
- GeneReview/NIH/UW entry on Holoprosencephaly Overview
- holoprosencephaly at NINDS
- http://www.familiesforhope.org
- http://www.holoprosencephaly.net
Congenital malformations and deformations of nervous system (Q00–Q07, 740–742) Brain Anencephaly (Acephaly, Acrania, Acalvaria, Iniencephaly) · Encephalocele · Arnold–Chiari malformationOtherCNS cyst (Porencephaly, Schizencephaly)Spinal cord OtherCategories:- Congenital disorders of nervous system

Wikimedia Foundation. 2010.