- Ligand (biochemistry)
-
For other uses of "Ligand", see Ligand (disambiguation).
In biochemistry and pharmacology, a ligand (from the Latin ligandum, binding) is a substance that forms a complex with a biomolecule to serve a biological purpose. In a narrower sense, it is a signal triggering molecule, binding to a site on a target protein.
The binding occurs by intermolecular forces, such as ionic bonds, hydrogen bonds and van der Waals forces. The docking (association) is usually reversible (dissociation). Actual irreversible covalent binding between a ligand and its target molecule is rare in biological systems. In contrast to the meaning in metalorganic and inorganic chemistry, it is irrelevant whether the ligand actually binds at a metal site, as is the case in hemoglobin.
Ligand binding to a receptor alters the chemical conformation, which is the three dimensional shape of the receptor protein. The conformational state of a receptor protein determines the functional state of a receptor. Ligands include substrates, inhibitors, activators, and neurotransmitters. The tendency or strength of binding is called affinity.
Radioligands are radioisotope labeled compounds and used in vivo as tracers in PET studies and for in vitro binding studies.
Contents
Receptor/ligand binding affinity
The interaction of most ligands with their binding sites can be characterized in terms of a binding affinity. In general, high affinity ligand binding results from greater intermolecular force between the ligand and its receptor while low affinity ligand binding involves less intermolecular force between the ligand and its receptor. In general, high affinity binding involves a longer residence time for the ligand at its receptor binding site than is the case for low affinity binding. High affinity binding of ligands to receptors is often physiologically important when some of the binding energy can be used to cause a conformational change in the receptor, resulting in altered behavior of an associated ion channel or enzyme.
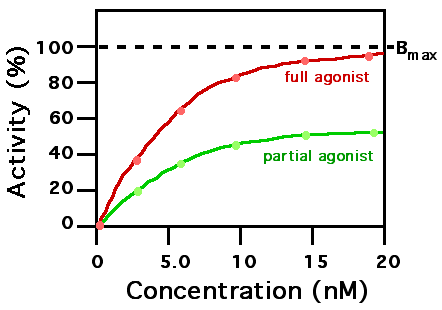
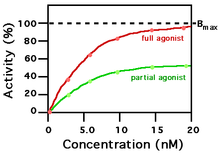 Two agonists with similar binding affinity
Two agonists with similar binding affinity
A ligand that can bind to a receptor, alter the function of the receptor and trigger a physiological response is called an agonist for that receptor. Agonist binding to a receptor can be characterized both in terms of how much physiological response can be triggered and in terms of the concentration of the agonist that is required to produce the physiological response. High-affinity ligand binding implies that a relatively low concentration of a ligand is adequate to maximally occupy a ligand-binding site and trigger a physiological response. Low-affinity binding implies that a relatively high concentration of a ligand is required before the binding site is maximally occupied and the maximum physiological response to the ligand is achieved. In the example shown to the right, two different ligands bind to the same receptor binding site. Only one of the agonists shown can maximally stimulate the receptor and, thus, can be defined as a "full agonist". An agonist that can only partially activate the physiological response is called a "partial agonist". Ligands that bind to a receptor but fail to activate the physiological response are receptor "antagonists". In this example, the concentration at which the full agonist (red curve) can half-maximally activate the receptor is about 5 x 10−9 Molar (nM = nanomolar).
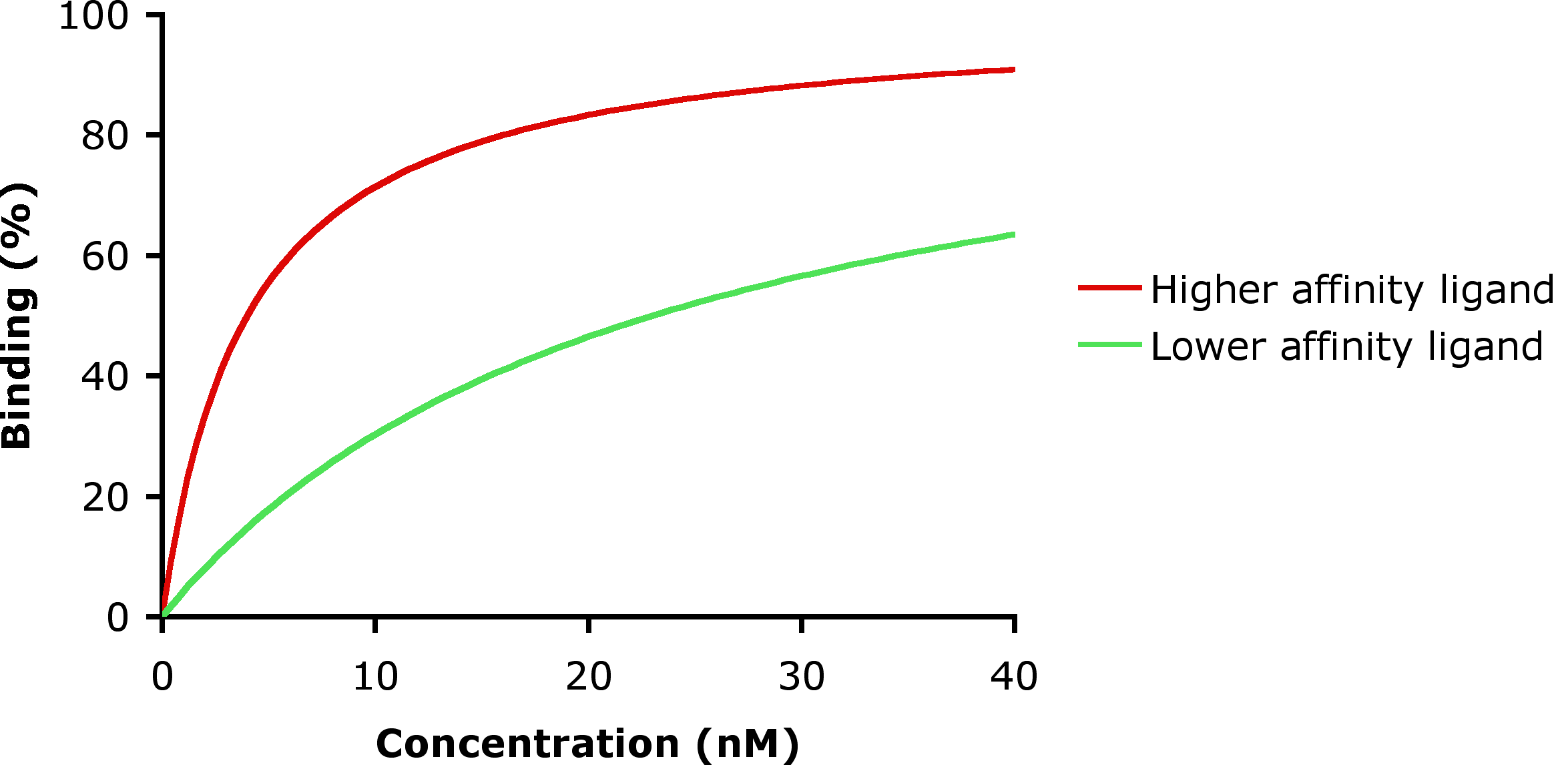
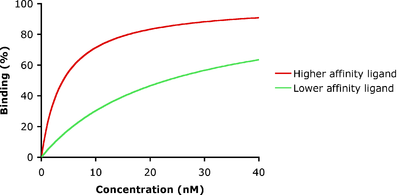 Two ligands with different receptor binding affinity.
Two ligands with different receptor binding affinity.In the example shown to the left, ligand-binding curves are shown for two ligands with different binding affinities. Ligand binding is often characterized in terms of the concentration of ligand at which half of the receptor binding sites are occupied, known as the dissociation constant (Kd). The ligand illustrated by the red curve has a higher binding affinity and smaller Kd than the ligand illustrated by the green curve. If these two ligands were present at the same time, more of the higher-affinity ligand would be bound to the available receptor binding sites. This is how carbon monoxide can compete with oxygen in binding to hemoglobin, resulting in carbon monoxide poisoning.
Binding affinity is most commonly determined using a radiolabeled ligand, known as hot ligand. Homologous competitive binding experiments involve binding-site competition between a hot ligand and a cold ligand (untagged ligand).[1] Non labelled methods such as surface plasmon resonance and dual polarisation interferometry can also quantify the affinity from concentration based assays but also from the kinetics of association and dissociation, and, in the later case, the conformational change induced upon binding. Recently, Microscale Thermophoresis (MST), an immobilization-free method[2] was developed. This method allows to determine the binding affinity without any limitation to the ligand's molecular weight.[3]
For the use of statistical mechanics in a quantitative study of the ligand-receptor binding affinity, see the comprehensive article[4] on the configuration integral.
Drug potency and binding affinity
Binding affinity data alone does not determine the overall potency of a drug. Potency is a result of the complex interplay of both the binding affinity and the ligand efficacy. Ligand efficacy refers to the ability of the ligand to produce a biological response upon binding to the target receptor and the quantitative magnitude of this response. This response may be as an agonist, antagonist, or inverse agonist, depending on the physiological response produced.[5]
Selective and non-selective
Selective ligands have a tendency to bind to a very limited types of receptors, whereas non-selective ligands bind to several types of receptors. This plays an important role in pharmacology, where drugs that are non-selective tend to have more adverse effects, because they bind to several other receptors in addition to the one generating the desired effect.
See also
External links
- BindingDB, a public database of measured protein-ligand binding affinities.
References
- ^ See Homologous competitive binding curves, A complete guide to nonlinear regression, curvefit.com.
- ^ Baaske P, Wienken CJ, Reineck P, Duhr S, Braun D (Feb 2010). "Optical Thermophoresis quantifies Buffer dependence of Aptamer Binding". Angew. Chem. Int. Ed. 49 (12): 1–5. doi:10.1002/anie.200903998. PMID 20186894. Lay summary – Phsyorg.com.
- ^ Wienken CJ et al. (2010). "Protein-binding assays in biological liquids using microscale thermophoresis.". Nature Communications 1 (7). Bibcode 2010NatCo...1E.100W. doi:10.1038/ncomms1093. http://www.nature.com/ncomms/journal/v1/n7/full/ncomms1093.html.
- ^ Vu-Quoc, L., Configuration integral (statistical mechanics), 2008.
- ^ Kenakin, Terrance P. (November 2006). A pharmacology primer: theory, applications, and methods. Academic Press. p. 79. ISBN 978-0123705990. http://books.google.com/books?id=d0phlW5cCmQC&lpg=PA79&dq=potency%20binding%20affinity%20agonist&lr&pg=PA79#v=onepage&q&f=false. "There are two main differences between binding and functional experiments: Functional responses are usually highly amplified translations of receptor stimulus (see Chapter 2). Therefore, while binding signals emanate from complete receptor populations, functional readouts often utilize only a small fraction of the receptor population in the preparation. This can lead to a greatly increased sensitivity to drugs that possess efficacy. No differences should be seen for antagonists. This amplification can be especially important for the detection of agonism, since potency may be more a function of ligand efficacy than affinity. Thus, a highly efficacious agonist may produce detectable responses at 100 to l,000 times lower concentrations than those that produce measurable amounts of displacement of a tracer in binding studies. The complex interplay between affinity and efficacy can be misleading in structure activity studies for agonists. For example. Figure 5.1 shows the lack of correlation of relative agonist potency for two dopamine receptor subtypes and the binding affinity on those receptor subtypes for a series of dopamine agonists. This data show that, for these molecules, changes in chemical structure lead to changes in relative efficacy not reflected in the affinity measurement. The relevant activity is relative agonist potency. Therefore, the affinity data are misleading. In this case, a functional assay is the correct approach for optimization of these molecules."
Signaling pathways Agents Receptor ligandsIntracellular signaling P+PsSignal transducing adaptor protein: Scaffold protein
2nd messenger: cAMP-dependent pathway · Ca2+ signaling · Lipid signaling · IP3/DAG pathwayBy location Other concepts B trdu: iter (nrpl/grfl/cytl/horl), csrc (lgic, enzr, gprc, igsr, intg, nrpr/grfr/cytr), itra (adap, gbpr, mapk), calc, lipd; path (hedp, wntp, tgfp+mapp, notp, jakp, fsap, hipp, tlrp) Intercellular signaling peptides and proteins / ligands Growth factors Endothelial growth factor • Epidermal growth factor • Fibroblast growth factor • Nerve growth factor • Platelet-derived growth factor • Transforming growth factor beta superfamilyEphrin Other Adipokine • Agouti signaling protein • Agouti-related protein • Angiogenic protein • CCN intercellular signaling protein (Cysteine-rich protein 61, Connective tissue growth factor, Nephroblastoma overexpressed protein) • Cytokine • Endothelin (EDN1, EDN2, EDN3) • Hedgehog protein • Interferon • Kinin • Parathyroid hormone-related protein • Semaphorin • Somatomedin • Tolloid-like metalloproteinase • Tumor necrosis factor • Wnt proteinsee also extracellular ligand disorders
B trdu: iter (nrpl/grfl/cytl/horl), csrc (lgic, enzr, gprc, igsr, intg, nrpr/grfr/cytr), itra (adap, gbpr, mapk), calc, lipd; path (hedp, wntp, tgfp+mapp, notp, jakp, fsap, hipp, tlrp)Categories:- Biomolecules
- Cell signaling
- Chemical bonding
- Proteins
- Ligands
Wikimedia Foundation. 2010.