- Docking (molecular)
-
Docking glossary • Receptor or host or lock – The "receiving" molecule, most commonly a protein or other biopolymer. • Ligand or guest or key – The complementary partner molecule which binds to the receptor. Ligands are most often small molecules but could also be another biopolymer. • Docking – Computational simulation of a candidate ligand binding to a receptor. • Binding mode – The orientation of the ligand relative to the receptor as well as the conformation of the ligand and receptor when bound to each other. • Pose – A candidate binding mode. • Scoring – The process of evaluating a particular pose by counting the number of favorable intermolecular interactions such as hydrogen bonds and hydrophobic contacts. • Ranking – The process of classifying which ligands are most likely to interact favorably to a particular receptor based on the predicted free-energy of binding. edit  Schematic diagram illustrating the docking of a small molecule ligand (brown) to a protein receptor (green) to produce a complex.
Schematic diagram illustrating the docking of a small molecule ligand (brown) to a protein receptor (green) to produce a complex.
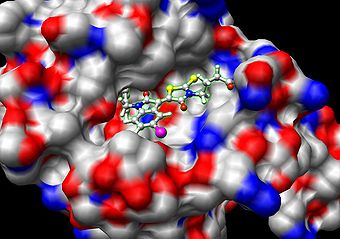
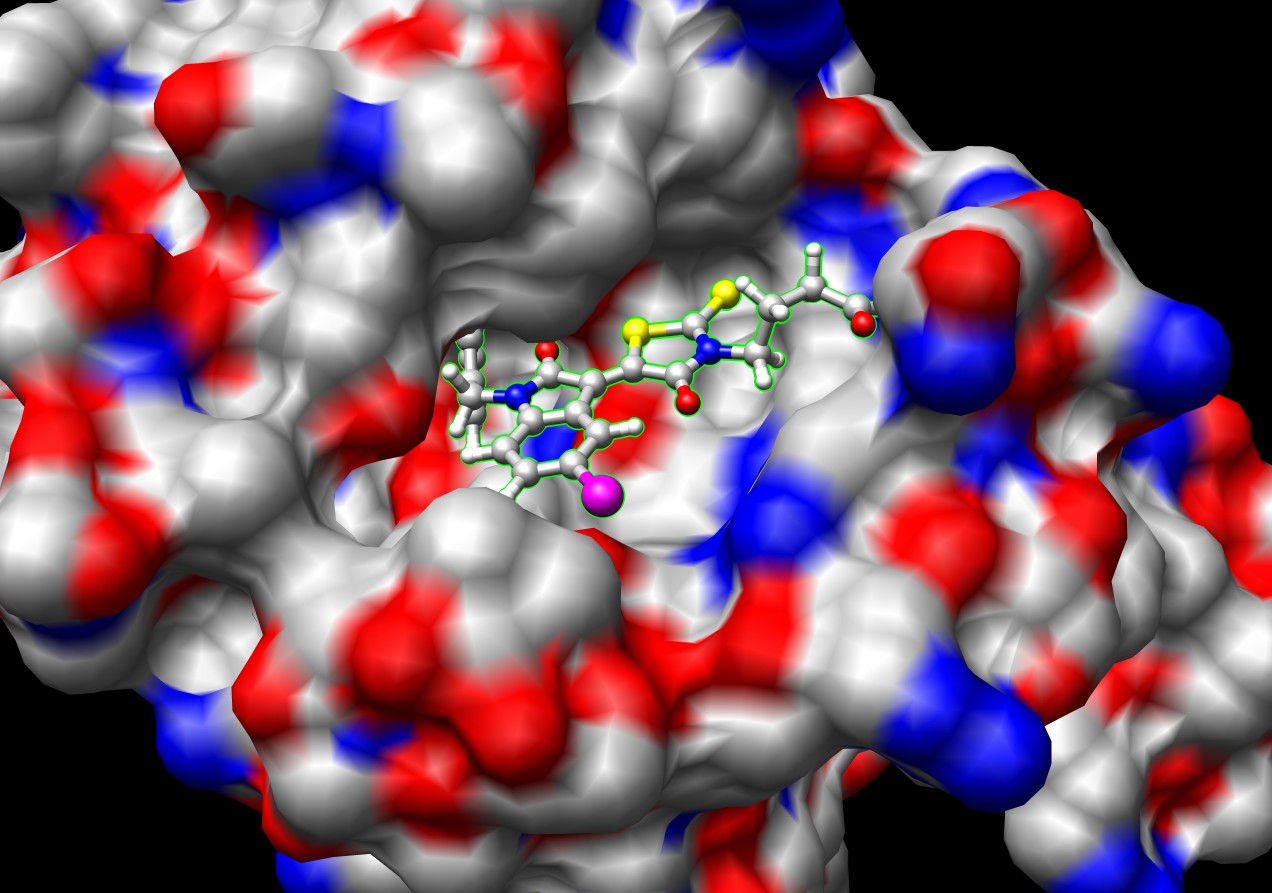
In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when bound to each other to form a stable complex.[1] Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using for example scoring functions.
The associations between biologically relevant molecules such as proteins, nucleic acids, carbohydrates, and lipids play a central role in signal transduction. Furthermore, the relative orientation of the two interacting partners may affect the type of signal produced (e.g., agonism vs antagonism). Therefore docking is useful for predicting both the strength and type of signal produced.
Docking is frequently used to predict the binding orientation of small molecule drug candidates to their protein targets in order to in turn predict the affinity and activity of the small molecule. Hence docking plays an important role in the rational design of drugs.[2] Given the biological and pharmaceutical significance of molecular docking, considerable efforts have been directed towards improving the methods used to predict docking .
Contents
Definition of problem
Molecular docking can be thought of as a problem of “lock-and-key”, where one is interested in finding the correct relative orientation of the “key” which will open up the “lock” (where on the surface of the lock is the key hole, which direction to turn the key after it is inserted, etc.). Here, the protein can be thought of as the “lock” and the ligand can be thought of as a “key”. Molecular docking may be defined as an optimization problem, which would describe the “best-fit” orientation of a ligand that binds to a particular protein of interest. However, since both the ligand and the protein are flexible, a “hand-in-glove” analogy is more appropriate than “lock-and-key”.[3] During the course of the process, the ligand and the protein adjust their conformation to achieve an overall “best-fit” and this kind of conformational adjustments resulting in the overall binding is referred to as “induced-fit”.[4]
The focus of molecular docking is to computationally simulate the molecular recognition process. The aim of molecular docking is to achieve an optimized conformation for both the protein and ligand and relative orientation between protein and ligand such that the free energy of the overall system is minimized.
Docking approaches
Two approaches are particularly popular within the molecular docking community. One approach uses a matching technique that describes the protein and the ligand as complementary surfaces.[5][6] The second approach simulates the actual docking process in which the ligand-protein pairwise interaction energies are calculated.[7] Both approaches have significant advantages as well as some limitations. These are outlined below.
Shape complementarity
Geometric matching/ shape complementarity methods describe the protein and ligand as a set of features that make them dockable.[8] These features may include molecular surface/ complementary surface descriptors. In this case, the receptor’s molecular surface is described in terms of its solvent-accessible surface area and the ligand’s molecular surface is described in terms of its matching surface description. The complementarity between the two surfaces amounts to the shape matching description that may help finding the complementary pose of docking the target and the ligand molecules. Another approach is to describe the hydrophobic features of the protein using turns in the main-chain atoms. Yet another approach is to use a Fourier shape descriptor technique.[9][10][11] Whereas the shape complementarity based approaches are typically fast and robust, they cannot usually model the movements or dynamic changes in the ligand/ protein conformations accurately, although recent developments allow these methods to investigate ligand flexibility. Shape complementarity methods can quickly scan through several thousand ligands in a matter of seconds and actually figure out whether they can bind at the protein’s active site, and are usually scalable to even protein-protein interactions. They are also much more amenable to pharmacophore based approaches, since they use geometric descriptions of the ligands to find optimal binding.
Simulation
The simulation of the docking process as such is a much more complicated process. In this approach, the protein and the ligand are separated by some physical distance, and the ligand finds its position into the protein’s active site after a certain number of “moves” in its conformational space. The moves incorporate rigid body transformations such as translations and rotations, as well as internal changes to the ligand’s structure including torsion angle rotations. Each of these moves in the conformation space of the ligand induces a total energetic cost of the system, and hence after every move the total energy of the system is calculated. The obvious advantage of the method is that it is more amenable to incorporate ligand flexibility into its modeling whereas shape complementarity techniques have to use some ingenious methods to incorporate flexibility in ligands. Another advantage is that the process is physically closer to what happens in reality, when the protein and ligand approach each other after molecular recognition. A clear disadvantage of this technique is that it takes longer time to evaluate the optimal pose of binding since they have to explore a rather large energy landscape. However grid-based techniques as well as fast optimization methods have significantly ameliorated these problems.
Mechanics of docking
To perform a docking screen, the first requirement is a structure of the protein of interest. Usually the structure has been determined using a biophysical technique such as x-ray crystallography, or less often, NMR spectroscopy. This protein structure and a database of potential ligands serve as inputs to a docking program. The success of a docking program depends on two components: the search algorithm and the scoring function.
Search algorithm
Main article: Searching the conformational space for dockingThe search space in theory consists of all possible orientations and conformations of the protein paired with the ligand. However in practice with current computational resources, it is impossible to exhaustively explore the search space—this would involve enumerating all possible distortions of each molecule (molecules are dynamic and exist in an ensemble of conformational states) and all possible rotational and translational orientations of the ligand relative to the protein at a given level of granularity. Most docking programs in use account for a flexible ligand, and several attempt to model a flexible protein receptor. Each "snapshot" of the pair is referred to as a pose.
A variety of conformational search strategies have been applied to the ligand and to the receptor. These include:
- systematic or stochastic torsional searches about rotatable bonds
- molecular dynamics simulations
- genetic algorithms to "evolve" new low energy conformations
Ligand flexibility
Conformations of the ligand may be generated in the absence of the receptor and subsequently docked[12] or conformations may be generated on-the-fly in the presence of the receptor binding cavity [13], or with full rotational flexibility of every dihedral angle using fragment based docking [14]. Force field energy evaluation are most often used to select energetically reasonable conformations,[15] but knowledge-based methods have also been used.[16]
Receptor flexibility
Computational capacity has increased dramatically over the last decade making possible the use of more sophisticated and computationally intensive methods in computer-assisted drug design. However, dealing with receptor flexibility in docking methodologies is still a thorny issue. The main reason behind this difficulty is the large number of degrees of freedom that have to be considered in this kind of calculations. Neglecting it, however, leads to poor docking results in terms of binding pose prediction.[17]
Multiple static structures experimentally determined for the same protein in different conformations are often used to emulate receptor flexibility.[18] Alternatively rotamer libraries of amino acid side chains that surround the binding cavity may be searched to generate alternate but energetically reasonable protein conformations.[19][20]
Scoring function
Main article: Scoring functions for dockingThe scoring function takes a pose as input and returns a number indicating the likelihood that the pose represents a favorable binding interaction.
Most scoring functions are physics-based molecular mechanics force fields that estimate the energy of the pose; a low (negative) energy indicates a stable system and thus a likely binding interaction. An alternative approach is to derive a statistical potential for interactions from a large database of protein-ligand complexes, such as the Protein Data Bank, and evaluate the fit of the pose according to this inferred potential.
There are a large number of structures from X-ray crystallography for complexes between proteins and high affinity ligands, but comparatively fewer for low affinity ligands as the later complexes tend to be less stable and therefore more difficult to crystallize. Scoring functions trained with this data can dock high affinity ligands correctly, but they will also give plausible docked conformations for ligands that do not bind. This gives a large number of false positive hits, i.e., ligands predicted to bind to the protein that actually don't when placed together in a test tube.
One way to reduce the number of false positives is to recalculate the energy of the top scoring poses using (potentially) more accurate but computationally more intensive techniques such as Generalized Born or Poisson-Boltzmann methods.[7]
Applications
A binding interaction between a small molecule ligand and an enzyme protein may result in activation or inhibition of the enzyme. If the protein is a receptor, ligand binding may result in agonism or antagonism. Docking is most commonly used in the field of drug design — most drugs are small organic molecules, and docking may be applied to:
- hit identification – docking combined with a scoring function can be used to quickly screen large databases of potential drugs in silico to identify molecules that are likely to bind to protein target of interest (see virtual screening).
- lead optimization – docking can be used to predict in where and in which relative orientation a ligand binds to a protein (also referred to as the binding mode or pose). This information may in turn be used to design more potent and selective analogs.
- Bioremediation – Protein ligand docking can also be used to predict pollutants that can be degraded by enzymes.[21]
See also
- Drug design
- Katchalski-Katzir algorithm
- List of molecular graphics systems
- Macromolecular docking
- Molecular mechanics
- Protein structure
- Protein design
- Software for molecular mechanics modeling
- Molecular design software
- Docking@Home
- Ibercivis
- ZINC database
- AutoDock
- DOCK
- Lead Finder
References
- ^ Lengauer T, Rarey M (1996). "Computational methods for biomolecular docking". Curr. Opin. Struct. Biol. 6 (3): 402–6. doi:10.1016/S0959-440X(96)80061-3. PMID 8804827.
- ^ Kitchen DB, Decornez H, Furr JR, Bajorath J (2004). "Docking and scoring in virtual screening for drug discovery: methods and applications". Nature reviews. Drug discovery 3 (11): 935–49. doi:10.1038/nrd1549. PMID 15520816.
- ^ Jorgensen WL (1991). "Rusting of the lock and key model for protein-ligand binding". Science 254 (5034): 954–5. doi:10.1126/science.1719636. PMID 1719636.
- ^ Wei BQ, Weaver LH, Ferrari AM, Matthews BW, Shoichet BK (2004). "Testing a flexible-receptor docking algorithm in a model binding site". J. Mol. Biol. 337 (5): 1161–82. doi:10.1016/j.jmb.2004.02.015. PMID 15046985.
- ^ Meng EC, Shoichet BK, Kuntz ID (2004). "Automated docking with grid-based energy evaluation". Journal of Computational Chemistry 13 (4): 505–524. doi:10.1002/jcc.540130412.
- ^ Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998). "Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function". Journal of Computational Chemistry 19 (14): 1639–1662. doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B.
- ^ a b Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL (2004). "Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures". Journal of Computational Chemistry 25 (2): 265–84. doi:10.1002/jcc.10378. PMID 14648625.
- ^ Shoichet BK, Kuntz ID, Bodian DL (2004). "Molecular docking using shape descriptors". Journal of Computational Chemistry 13 (3): 380–397. doi:10.1002/jcc.540130311.
- ^ Cai W, Shao X, Maigret B (January 2002). "Protein-ligand recognition using spherical harmonic molecular surfaces: towards a fast and efficient filter for large virtual throughput screening". J. Mol. Graph. Model. 20 (4): 313–28. doi:10.1016/S1093-3263(01)00134-6. PMID 11858640.
- ^ Morris RJ, Najmanovich RJ, Kahraman A, Thornton JM (May 2005). "Real spherical harmonic expansion coefficients as 3D shape descriptors for protein binding pocket and ligand comparisons". Bioinformatics 21 (10): 2347–55. doi:10.1093/bioinformatics/bti337. PMID 15728116.
- ^ Kahraman A, Morris RJ, Laskowski RA, Thornton JM (April 2007). "Shape variation in protein binding pockets and their ligands". J. Mol. Biol. 368 (1): 283–301. doi:10.1016/j.jmb.2007.01.086. PMID 17337005.
- ^ Kearsley SK, Underwood DJ, Sheridan RP, Miller MD (October 1994). "Flexibases: a way to enhance the use of molecular docking methods". J. Comput. Aided Mol. Des. 8 (5): 565–82. doi:10.1007/BF00123666. PMID 7876901.
- ^ Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (March 2004). "Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy". J. Med. Chem. 47 (7): 1739–49. doi:10.1021/jm0306430. PMID 15027865.
- ^ Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP (July 2007). "eHiTS: A new fast, exhaustive flexible ligand docking system". Journal of Molecular Graphics and Modelling 26 (1): 198–212. doi:10.1016/j.jmgm.2006.06.002. PMID 16860582.
- ^ Wang Q, Pang YP (2007). Romesberg, Floyd. ed. "Preference of small molecules for local minimum conformations when binding to proteins". PLoS ONE 2 (9): e820. doi:10.1371/journal.pone.0000820. PMC 1959118. PMID 17786192. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1959118.
- ^ Klebe G, Mietzner T (October 1994). "A fast and efficient method to generate biologically relevant conformations". J. Comput. Aided Mol. Des. 8 (5): 583–606. doi:10.1007/BF00123667. PMID 7876902.
- ^ Cerqueira NM, Bras NF, Fernandes PA, Ramos MJ (January 2009). "MADAMM: a multistaged docking with an automated molecular modeling protocol". Proteins 74 (1): 192–206. doi:10.1002/prot.22146. PMID 18618708.
- ^ Totrov M, Abagyan R (April 2008). "Flexible ligand docking to multiple receptor conformations: a practical alternative". Curr. Opin. Struct. Biol. 18 (2): 178–84. doi:10.1016/j.sbi.2008.01.004. PMC 2396190. PMID 18302984. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2396190.
- ^ Hartmann C, Antes I, Lengauer T (February 2009). "Docking and scoring with alternative side-chain conformations". Proteins 74 (3): 712–26. doi:10.1002/prot.22189. PMID 18704939.
- ^ Taylor RD, Jewsbury PJ, Essex JW (October 2003). "FDS: flexible ligand and receptor docking with a continuum solvent model and soft-core energy function". J Comput Chem 24 (13): 1637–56. doi:10.1002/jcc.10295. PMID 12926007.
- ^ Suresh PS, Kumar A, Kumar R, Singh VP (January 2008). "An in silico [correction of insilico] approach to bioremediation: laccase as a case study". J. Mol. Graph. Model. 26 (5): 845–9. doi:10.1016/j.jmgm.2007.05.005. PMID 17606396.
External links
- Bikadi Z, Kovacs S, Demko L, Hazai E. "Molecular Docking Server - Ligand Protein Docking & Molecular Modeling". Virtua Drug Ltd. http://www.dockingserver.com. Retrieved 2008-07-15. "Internet service that calculates the site, geometry and energy of small molecules interacting with proteins"
- Malinauskas T. "Step by step installation of MGLTools 1.5.2 (AutoDockTools, Python Molecular Viewer and Visual Programming Environment) on Ubuntu Linux 8.04". http://users.ox.ac.uk/~jesu1458/installation_of_autodock_on_ubuntu_linux/. Retrieved 2008-07-15.
- Malinauskas T. "High-throughput molecular docking using free tools: ZINC 8, AutoDockTools 1.5.2 and Docker 1.0". http://users.ox.ac.uk/~jesu1458/docker/. Retrieved 2008-07-23.
- AutoDock and MGLTools for Debian
- Docking@GRID Project of Conformational Sampling and Docking on Grids : one aim is to deploy some intrinsic distributed docking algorithms on computational Grids, download Docking@GRID open-source Linux version
- Docking software
- Click2Drug.org - Directory of computational drug design tools.
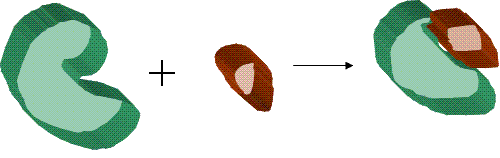
Wikimedia Foundation. 2010.