- Human mitochondrial genetics
-
Human mitochondrial genetics is the study of the genetics of the DNA contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.
Mitochondrial DNA (mtDNA) is not transmitted through nuclear DNA (nDNA). In humans, as in most multicellular organisms, mitochondrial DNA is inherited only from the mother's ovum.[1]
Mitochondrial inheritance is therefore non-Mendelian, as Mendelian inheritance presumes that half the genetic material of a fertilized egg (zygote) derives from each parent.
Eighty percent of mitochondrial DNA codes for functional mitochondrial proteins, and therefore most mitochondrial DNA mutations lead to functional problems, which may be manifested as muscle disorders (myopathies).
Understanding the genetic mutations that affect mitochondria can help us to understand the inner workings of cells and organisms, as well as helping to suggest methods for successful therapeutic tissue and organ cloning, and to treatments or possibly cures for many devastating muscular disorders.
Contents
Mitochondrial function and genome
Because they provide 36 molecules of ATP per glucose molecule in contrast to the 2 ATP molecules produced by glycolysis, mitochondria are essential to all higher organisms for sustaining life. The mitochondrial diseases are genetic disorders carried in mitochondrial DNA, or nuclear DNA coding for mitochondrial components. Slight problems with any one of the numerous enzymes used by the mitochondria can be devastating to the cell, and in turn, to the organism.
Membrane complexes
The processes carried out by the electron transport chain are mediated by protein complexes (named Complexes I-V, DHO-QO, ETF-QO, and ANT). Complex I, or NADH : coenzyme Q oxidoreductase, uses the energy in NADH to pump protons into the intermembrane space of the mitochondrion, pumping 2 protons per electron and passing 2 electrons via coenzyme Q to complex III or coenzyme Q : cytochrome c oxidoreductase. Complex II or succinate : coenzyme Q oxidoreductase accepts energy from succinate produced in the citric acid cycle and passes it via coenzyme Q to complex III. Complex III pumps 1 protons per electron and passes 1 electron via cytochrome c to complex IV or Cytochrome C : O2 Oxidoreductase. Complex IV pumps 1 protons into the space between the mitochondrion’s two membranes before passing the electron to O2 which reacts to form water. Complex V (ATP synthase) is a rotary complex which allows approximately (determining the actual number is very difficult) 10 protons to enter the mitochondrial matrix along their concentration gradients. It uses the energy from the gradient to form the bond between ADP and the phosphate group to create ATP. The electron transfer flavoprotein : coenzyme Q oxidoreductase is also an electron-transporting molecule and is involved in the breakdown of fatty acids and amino acids. ANT (adenine nucleotide translocator) is also involved in oxidative phosphorylation as an energy carrying molecule. Each of these eight complexes plays a vital role in the health of the cell and any slight mutation in any one of the proteins that make up these complexes can lead to cell death or stress, which can both in turn lead to a number of diseases.
Genome
Further information: Mitochondrial DNAMitochondrial DNA (mtDNA) is present in mitochondria as a circular molecule and in most species codes for 13 or 14 proteins involved in the electron transfer chain, 2 rRNA subunits and 22 tRNA molecules (all necessary for protein synthesis). The number of proteins involved in the electron transfer chain is much larger than 13 or 14, but the others are coded by the nuclear DNA.
In total, the mitochondrion hosts about 3000 proteins, but only about 13 of them are coded on the mitochondrial DNA. Most of the 3000 proteins are involved in a variety of processes other than ATP production, such as porphyrin synthesis. Only about 3% of them code for ATP production proteins. This means most of the genetic information coding for the protein makeup of mitochondria is in chromosomal DNA and is involved in processes other than ATP synthesis. This increases the chances that a mutation that will affect a mitochondrion will occur in chromosomal DNA, which is inherited in a Mendelian pattern. Another result is that a chromosomal mutation will affect a specific tissue due to its specific needs, whether those may be high energy requirements or a need for the catabolism or anabolism of a specific neurotransmitter or nucleic acid. Because several copies of the mitochondrial genome are carried by each mitochondrion (2-10 in humans), mitochondrial mutations can be inherited maternally by mtDNA mutations which are present in mitochondria inside the oocyte before fertilization, or (as stated above) through mutations in the chromosomes.
In humans, the heavy strand of mtDNA carries 28 genes and the light strand of mtDNA carries only 9 genes. Eight of the 9 genes on the light strand code for mitochondrial tRNA molecules. Human mtDNA consists of 16,569 nucleotide pairs. The entire molecule is regulated by only one regulatory region which contains the origins of replication of both heavy and light strands. The entire human mitochondrial DNA molecule has been mapped[1][2]. The rate of mutation in mtDNA is calculated to be about ten times greater than that of nuclear DNA, possibly due to a paucity of DNA repair mechanisms. This high mutation rate leads to a high variation between mitochondria, not only among different species but even within the same species. It is calculated that if two humans are chosen randomly and their mtDNA is tested, they will have an average of between fifty and seventy different nucleotides. This may not seem like much, but when compared to the total number of nucleotides of a human mitochondrial DNA molecule (16,569), as much as 0.42% of the mtDNA varies between two people.
Genetic code variants
The genetic code is, for the most part, universal, with few exceptions: mitochondrial genetics includes some of these. For most organisms the "stop codons" are “UAA”, “UAG”, and “UGA”. In vertebrate mitochondria “AGA” and “AGG” are also stop codons, but not “UGA”, which codes for tryptophan instead. “AUA” codes for isoleucine in most organisms but for methionine in vertebrate mitochondrial mRNA.
There are many other variations among the codes used by other mitochondrial m/tRNA, which happened not to be harmful to their organisms, and which can be used as a tool (along with other mutations among the mtDNA/RNA of different species) to determine relative proximity of common ancestry of related species. (The more related two species are, the more mtDNA/RNA mutations will be the same in their mitochondrial genome).
Using these techniques, it is estimated that the first mitochondria arose around 1.5 billion years ago. A generally accepted hypothesis is that mitochondria originated as an aerobic prokaryote in a symbiotic relationship within an anaerobic eukaryote.
Inheritance patterns
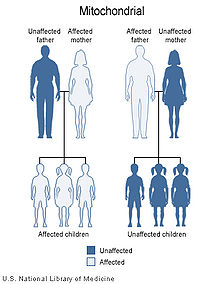 Mitochondrial Inheritance Patterns
Mitochondrial Inheritance Patterns
Because mitochondrial diseases (diseases due to malfunction of mitochondria) can be inherited both maternally and through chromosomal inheritance, the way in which they are passed on from generation to generation can vary greatly depending on the disease. Mitochondrial genetic mutations that occur in the nuclear DNA can occur in any of the chromosomes (depending on the species). Mutations inherited through the chromosomes can be autosomal dominant or recessive and can also be sex-linked dominant or recessive. Chromosomal inheritance follows normal Mendelian laws, despite the fact that the phenotype of the disease may be masked.
Because of the complex ways in which mitochondrial and nuclear DNA "communicate" and interact, even seemingly simple inheritance is hard to diagnose. A mutation in chromosomal DNA may change a protein that regulates (increases or decreases) the production of another certain protein in the mitochondria or the cytoplasm; this may lead to slight, if any, noticeable symptoms. On the other hand, some devastating mtDNA mutations are easy to diagnose because of their widespread damage to muscular, neural, and/or hepatic tissues (among other high-energy and metabolism-dependent tissues) and because they are present in the mother and all the offspring.
Mitochondrial genome mutations are passed on 100% of the time from mother to all her offspring. The number of affected mtDNA molecules inherited by a specific offspring can vary greatly because
- the mitochondria within the fertilized oocyte is what the new life will have to begin with (in terms of mtDNA),
- the number of affected mitochondria varies from cell (in this case, the fertilized oocyte) to cell depending both on the number it inherited from its mother cell and environmental factors which may favor mutant or wildtype mitochondrial DNA,
- the number of mtDNA molecules in the mitochondria varies from around two to ten.
It is possible, even in twin births, for one baby to receive more than half mutant mtDNA molecules while the other twin may receive only a tiny fraction of mutant mtDNA molecules with respect to wildtype (depending on how the twins divide from each other and how many mutant mitochondria happen to be on each side of the division). In a few cases, some mitochondria or a mitochondrion from the sperm cell enters the oocyte but paternal mitochondria are actively decomposed.
Replication, repair, transcription, and translation
Mitochondrial replication is controlled by nuclear genes and is specifically suited to make as many mitochondria as that particular cell needs at the time. Human mitochondrial DNA (mtDNA) has three promoters, H1, H2, and L (heavy strand 1, heavy strand 2, and light strand promoters). The H2 promoter transcribes almost the entire heavy strand and the L promoter transcribes the entire light strand. The H1 promoter causes the transcription of the two mitochondrial rRNA molecules. When transcription takes place on the heavy strand a polycistronic transcript is created. The light strand produces either small transcripts, which can be used as primers, or one long transcript. The production of primers occurs by processing of light strand transcripts with the Mitochondrial RNase MRP (Mitochondrial RNA Processing). The requirement of transcription to produce primers links the process of transcription to mtDNA replication. Full length transcripts are cut into functional tRNA, rRNA, and mRNA molecules. The process of transcription initiation in mitochondria involves three types of proteins: the mitochondrial RNA polymerase (POLRMT), mitochondrial transcription factor A (TFAM), and mitochondrial transcription factors B1 and B2 (TFB1M, TFB2M). POLRMT, TFAM, and TFB1M or TFB2M assemble at the mitochondrial promoters and begin transcription. The actual molecular events that are involved in initiation are unknown, but these factors make up the basal transcription machinery and have been shown to function in vitro. Mitochondrial translation is still not very well understood. In vitro translations have still not been successful, probably due to the difficulty of isolating sufficient mt mRNA, functional mt rRNA, and possibly because of the complicated changes that the mRNA undergoes before it is translated.
Mitochondrial DNA polymerase
The Mitochondrial DNA Polymerase (Pol gamma, encoded by the POLG gene) is used in the copying of mtDNA during replication. Because the two (heavy and light) strands on the circular mtDNA molecule have different origins of replication, it replicates in a D-loop mode. One strand begins to replicate first, displacing the other strand. This continues until replication reaches the origin of replication on the other strand, at which point the other strand begins replicating in the opposite direction. This results in two new mtDNA molecules. Each mitochondrion has several copies of the mtDNA molecule and the number of mtDNA molecules is a limiting factor in mitochondrial fission. After the mitochondrion has enough mtDNA, membrane area, and membrane proteins, it can undergo fission (very similar to that which bacteria use) to become two mitochondria. Evidence suggests that mitochondria can also undergo fusion and exchange (in a form of crossover) genetic material among each other. Mitochondria sometimes form large matrices in which fusion, fission, and protein exchanges are constantly occurring. mtDNA shared among mitochondria (despite the fact that they can undergo fusion).
Damage and transcription error
Mitochondrial DNA is susceptible to damage from free oxygen radicals from mistakes that occur during the production of ATP through the electron transport chain. These mistakes can be caused by genetic disorders, cancer, and temperature variations. These radicals can damage mtDNA molecules or change them, making it hard for mitochondrial polymerase to replicate them. Both cases can lead to deletions, rearrangements, and other mutations. Recent evidence has suggested that mitochondria have enzymes that proofread mtDNA and fix mutations that may occur due to free radicals. It is believed that a DNA recombinase found in mammalian cells is also involved in a repairing recombination process. Deletions and mutations due to free radicals have been associated with the aging process. It is believed that radicals cause mutations which lead to mutant proteins, which in turn lead to more radicals. This process takes many years and is associated with some aging processes involved in oxygen-dependent tissues such as brain, heart, muscle, and kidney. Auto-enhancing processes such as these are possible causes of degenerative diseases including Parkinson’s, Alzheimer’s, and coronary artery disease.
Chromosomally mediated mtDNA replication errors
Because mitochondrial growth and fission are mediated by the nuclear DNA, mutations in nuclear DNA can have a wide array of effects on mtDNA replication. Despite the fact that the loci for some of these mutations have been found on human chromosomes, specific genes and proteins involved have not yet been isolated. Mitochondria need a certain protein to undergo fission. If this protein (made by the nucleus) is not present, the mitochondria grow but they do not divide. This leads to giant, inefficient mitochondria. Mistakes in chromosomal genes or their products can also affect mitochondrial replication more directly by inhibiting mitochondrial polymerase and can even cause mutations in the mtDNA directly and indirectly. Indirect mutations are most often caused by radicals created by defective proteins made from nuclear DNA.
Mitochondrial diseases
Mitochondrial diseases range in severity from asymptomatic to fatal, and are most commonly due to inherited rather than acquired mutations of mitochondrial DNA. A given mitochondrial mutation can cause various diseases depending on the severity of the problem in the mitochondria and the tissue the affected mitochondria are in. Conversely, several different mutations may present themselves as the same disease. This almost patient-specific characterization of mitochondrial diseases (see Personalized medicine) makes them very hard to accurately recognize, diagnose and trace. Some diseases are observable at or even before birth (many causing death) while others do not show themselves until late adulthood (late-onset disorders). This is because the number of mutant versus wildtype mitochondria varies between cells and tissues, and is continuously changing. Because cells have multiple mitochondria, different mitochondria in the same cell can have different variations of the mtDNA. This condition is referred to as heteroplasmy. When a certain tissue reaches a certain ratio of mutant versus wildtype mitochondria, a disease will present itself. The ratio varies from person to person and tissue to tissue (depending on its specific energy, oxygen, and metabolism requirements, and the effects of the specific mutation). Mitochondrial diseases are very numerous and different. Apart from diseases caused by abnormalities in mitochondrial DNA, many diseases are suspected to be associated in part by mitochondrial dysfunctions, such as diabetes mellitus, forms of cancer and cardiovascular disease, lactic acidosis, specific forms of myopathy, osteoporosis, Alzheimer's disease, Parkinsons's disease, stroke, Male infertility and which are also believed to play a role in the aging process.
See also
- Paternal mtDNA transmission
- Human mitochondrial DNA haplogroups
- Cambridge Reference Sequence
- Human mitochondrial molecular clock
- Genetic genealogy for lists of databases which help users find others with their Y-DNA and mtDNA.
Notes
- ^ "Societat Catalana de Neurologia". http://www.scn.es/cursos/muscular/genmito/mitocondria.htm. Retrieved December 5 2005.
- ^ "http://www.mitomap.org/mitomap/mitomapgenome.pdf" (PDF). http://www.mitomap.org/mitomapgenome.pdf. Retrieved December 5 2005.
References
- Roberts D, Frerman F, and Kim J. (December 1996) Three-dimensional Structure of Human Electron Transfer Flavoprotein to 2.1-Å Resolution. Proc. Natl. Acad. Sci. USA. Vol. 93. (dl November 2004).
- Naviaux, Robert. (1997) The Spectrum of Mitochondrial Disease. Exceptional Parent Magazine. Vol. 27, Issue 8.
- Crofts, Antony R. (1996) Biophysics 354: Biological Energy Conversions. University of Illinois at Urbana-Champaign. (dl November 2004).
- Nester, Anderson, Roberts, Pearsall, Nester. Microbiology, A Human Perspective. Boston: McGraw Hill Inc; 2004.
- Mitochondrial inheritance tree Mitochondrial DNA genetics
- Broughton R, Milam J, and Roe B. (November 2001) The Complete Sequence of the Zebrafish (Danio rerio) Mitochondrial Genome and Evolutionary Patterns in Vertebrate Mitochondrial DNA. Genome Research. Vol. 11, Issue 11. (dl November 2004).
- Neuromuscular Disease Center. Washington University, St. Louis, MO; October 10, 2004. (dl November 2004).
- Montoya J, Playán A, Alcaine M, Enríquez J, Fernández-Silva P, López-Pérez M, Martinez-Azorín F, Pérez-Martos A. Societat Catalana de Neurologia. Universidad de Zaragoza. (dl November 2004).
- United Mitochondrial Disease Foundation. Pittsburgh, PA. (dl November 2004).
- Tamarin, Robert H. Principles of Genetics, Seventh Edition. New York, NY: McGraw-Hill Inc; 2002.
- Scheffler, Immo E. (2000) Review Article: A Century of Mitochondrial Research: Achievements and Perspectives. Elsevier Science B. V. and Mitochondrial Research Society. (dl November 2004).
- Vladutiu, Georgirene D. (1997) Advances in Mitochondrial Disease Research. Exceptional Parent Magazine. Vol. 27, Issue 8.
- DiMauro, Salvatore. (1997) Promising Avenues of Investigation in the Diagnosis and Treatment of Mitochondrial Defects. Exceptional Parent Magazine. Vol. 27, Issue 8.
- University of Texas Medical Branch. TX; December 2003. The Mitochondrial Life Cycle. (dl November 2004).
- The Mitochondrial Research Society. Mitochondria Means the Most to US; 2004. (dl November 2004).
- Purves W, Sadava D, Orians G, Heller C. Life: The Science of Biology, Sixth Edition. Massachusetts: Sinauer Associates, Inc; 2002.
- Venkatesh S, Deecaraman M, Kumar R, Shamsi MB, Dada R. Role of reactive oxygen species in the pathogenesis of mitochondrial DNA (mtDNA) mutations in male infertility. Indian J Med Res. 2009 Feb;129(2):127-37. Review.
Categories:
Wikimedia Foundation. 2010.