- Rotational spectroscopy
-
 Part of the rotational-vibrational spectrum of carbon monoxide (CO) gas (from FTIR), showing the presence of P- and R- branches. Frequency is on the x-axis, and absorbance on the y-axis.
Part of the rotational-vibrational spectrum of carbon monoxide (CO) gas (from FTIR), showing the presence of P- and R- branches. Frequency is on the x-axis, and absorbance on the y-axis.
Rotational spectroscopy or microwave spectroscopy studies the absorption and emission of electromagnetic radiation (typically in the microwave region of the electromagnetic spectrum) by molecules associated with a corresponding change in the rotational quantum number of the molecule. The use of microwaves in spectroscopy essentially became possible due to the development of microwave technology for RADAR during World War II. Rotational spectroscopy is practical only in the gas phase where the rotational motion is quantized. In solids or liquids the rotational motion is usually quenched due to collisions.
Rotational spectrum from a molecule (to first order) requires that the molecule have a dipole moment and that there be a difference between its center of charge and its center of mass, or equivalently a separation between two unlike charges. It is this dipole moment that enables the electric field of the light (microwave) to exert a torque on the molecule, causing it to rotate more quickly (in excitation) or slowly (in de-excitation). Homonuclear diatomic molecules such as dioxygen (O2), dihydrogen (H2), etc. do not have a dipole moment and, hence, no purely rotational spectrum. However, electronic excitations can lead to asymmetric charge distributions and thus provide a net dipole moment to the molecule. Under such circumstances, these molecules will exhibit a rotational spectrum.
Among the diatomic molecules, carbon monoxide (CO) has one of the simplest rotational spectra. As for tri-atomic molecules, hydrogen cyanide (HC≡N) has a simple rotational spectrum for a linear molecule and hydrogen isocyanide (HN=C:) for a non-linear molecule. As the number of atoms increases, the spectrum becomes more complex, as lines, due to different transitions, start overlapping.
Contents
Understanding the rotational spectrum
In quantum mechanics the free rotation of a molecule is quantized, that is the rotational energy and the angular momentum can take only certain fixed values; what these values are is related simply to the moment of inertia, I, of the molecule. In general, for any molecule, there are three moments of inertia: IA, IB and IC about three mutually orthogonal axes A, B, and C with the origin at the center of mass of the system. Linear molecules are a special case because one of the moments of inertia (IA, the moment of inertia along molecular axis) is negligible (i.e.,
 ).
).The general convention is to define the axes such that the axis A has the smallest moment of inertia, the axis B axis is the next smallest and so on such that
 . (A rarely used convention defines the A axis along a symmetric axis of the molecule, if one exists. To avoid confusion, this article uses the prior convention.) The particular pattern of energy levels (and, hence, of transitions in the rotational spectrum) for a molecule is determined by its symmetry. A convenient way to look at the molecules is to divide them into four different classes (based on the symmetry of their structure). These are linear molecules, symmetric tops, spherical tops, and asymmetric tops. (Equivalently, many authors use the word "rotor" instead of "top".)
. (A rarely used convention defines the A axis along a symmetric axis of the molecule, if one exists. To avoid confusion, this article uses the prior convention.) The particular pattern of energy levels (and, hence, of transitions in the rotational spectrum) for a molecule is determined by its symmetry. A convenient way to look at the molecules is to divide them into four different classes (based on the symmetry of their structure). These are linear molecules, symmetric tops, spherical tops, and asymmetric tops. (Equivalently, many authors use the word "rotor" instead of "top".)Linear molecules
For a linear molecule the moments of inertia are related by
. For most of the purposes, IA is taken to be zero. For a linear molecule, the separation of lines in the rotational spectrum can be related directly to the moment of inertia of the molecule, and, for a molecule of known atomic masses, can be used to determine the bond lengths (structure) directly. For diatomic molecules, this process is trivial, and can be made from a single measurement of the rotational spectrum. For linear molecules with more atoms, rather more work is required, and it is necessary to measure molecules in which more than one isotope of each atom have been substituted (effectively this gives rise to a set of simultaneous equations that can be solved for the bond lengths).Examples of linear molecules: dioxygen (O=O), carbon monoxide (O≡C*), hydroxy radical (OH), carbon dioxide (O=C=O), hydrogen cyanide (HC≡N), carbonyl sulfide (O=C=S), chloroethyne (HC≡CCl), acetylene (HC≡CH)
Symmetric tops
A symmetric top is a molecule in which two moments of inertia are the same. As a matter of historical convenience, spectroscopists divide molecules into two classes of symmetric tops, Oblate symmetric tops (saucer or disc shaped) with IA = IB < IC and Prolate symmetric tops (rugby football, or cigar shaped) with IA < IB = IC. The spectra look rather different, and are instantly recognizable. As with linear molecules, the structure of symmetric tops (bond lengths and bond angles) can be deduced from their spectra.
Examples of symmetric tops:
- Oblate: benzene (C6H6), cyclobutadiene (C4H4), ammonia (NH3)
- Prolate: chloromethane (CH3Cl), propyne (CH3C≡CH)
Spherical tops
A spherical top molecule can be considered as a special case of symmetric tops with equal moment of inertia about all three axes (IA = IB = IC).
Examples of spherical tops: edit] Asymmetric tops
An asymmetric top occurs if all three moments of inertia are different. Most large molecules are asymmetric tops, even when they have a high degree of symmetry. In general, for such molecules, a simple interpretation of the spectrum is not normally possible. However, molecules have spectra similar to those of a linear molecule or a symmetric top, in which case the molecular structure may also be similar to that of a linear molecule or a symmetric top. For the most general case, however, all that can be done is to fit the spectra to three different moments of inertia. If the molecular formula is known, then educated guesses can be made of the possible structure, and, from this guessed structure, the moments of inertia can be calculated. If the calculated moments of inertia agree well with the measured moments of inertia, then the structure can be said to have been determined. For this approach to determining molecular structure, isotopic substitution is invaluable.
Examples of asymmetric tops: anthracene (C14H10), water (H2O), nitrogen dioxide (NO2)
Structure of rotational spectra
Linear molecules
 An energy level diagram showing some of the transitions involved in the IR vibration-rotation spectrum of a linear molecule: P branch (where ΔJ = − 1), Q branch (not always allowed, ΔJ = 0) and R branch (ΔJ = 1)
An energy level diagram showing some of the transitions involved in the IR vibration-rotation spectrum of a linear molecule: P branch (where ΔJ = − 1), Q branch (not always allowed, ΔJ = 0) and R branch (ΔJ = 1)These molecules have two degenerate modes of rotation (IB = IC, IA = 0). Since we cannot distinguish between the two modes, we need only one rotational quantum number (J) to describe the rotational motion of the molecule.
The rotational energy levels (F(J)) of the molecule based on rigid rotor model can be expressed as,
where
 is the rotational constant of the molecule and is related to the moment of inertia of the molecule IB = IC by
is the rotational constant of the molecule and is related to the moment of inertia of the molecule IB = IC by(in wavenumber scale).
Selection rules dictate that during emission or absorption the rotational quantum number has to change by unity; i.e.,
 . Thus, the locations of the lines in a rotational spectrum will be given by
. Thus, the locations of the lines in a rotational spectrum will be given bywhere
 denotes the lower energy level and
denotes the lower energy level and  denotes higher energy level involved in the transition. The height of the lines is determined by the distribution of the molecules in the different levels and the probability of transition between two energy levels.
denotes higher energy level involved in the transition. The height of the lines is determined by the distribution of the molecules in the different levels and the probability of transition between two energy levels.We observe that, for a rigid rotor, the transition lines are equally spaced in the wavenumber space. However, this is not always the case, except for the rigid rotor model. For non-rigid rotor model, we need to consider changes in the moment of inertia of the molecule. Two primary reasons for this are
Centrifugal distortion
When a molecule rotates, the centrifugal force pulls the atoms apart. As a result, the moment of inertia of the molecule increases, thus decreasing
. To account for this a centrifugal distortion correction term is added to the rotational energy levels of the molecule.where
 is the centrifugal distortion constant.
is the centrifugal distortion constant.Therefore, the line spacing for the rotational mode changes to,
Effect of vibration on rotation
A molecule is always in vibration. As the molecule vibrates, its moment of inertia changes. Further, there is a fictitious force, Coriolis coupling, between the vibrational motion of the nuclei in the rotating (non-inertial) frame. However, as long as the vibrational quantum number does not change (i.e., the molecule is in only one state of vibration), the effect of vibration on rotation is not important, because the time for vibration is much shorter than the time required for rotation. The Coriolis coupling is often negligible, too, if one is interested in low vibrational and rotational quantum numbers only.
Symmetric top
The rotational motion of a symmetric top molecule can be described by two independent rotational quantum numbers (since two axes have equal moments of inertia, the rotational motion about these axes requires only one rotational quantum number for complete description). Instead of defining the two rotational quantum numbers for two independent axes, we associate one of the quantum number (J) with the total angular momentum of the molecule and the other quantum number (K) with the angular momentum of the axis that has different moment of inertia (i.e., axis C for oblate symmetric top and axis A for prolate symmetric tops). The rotational energy F(J,K) of such a molecule, based on rigid rotor assumptions can be expressed in terms of the two previously defined rotational quantum numbers as follows
where
 and
and  for a prolate symmetric top molecule or
for a prolate symmetric top molecule or  for an oblate molecule.
for an oblate molecule.Selection rule for these molecules provide the guidelines for possible transitions. Therefore,
 .
.
This is so because K is associated with the axis about which the molecule is symmetric and, hence, has no net dipole moment in that direction. Thus, there is no interaction of this mode with the light particles (photons).
This gives the transition wavenumbers of
which is the same as in the case of a linear molecule.
In case of non-rigid rotors, the first order centrifugal distortion correction is given by
The suffixes on the centrifugal distortion constant D indicate the rotational mode involved and are not a function of the rotational quantum number. The location of the transition lines on a spectrum is given by
Spherical top
Unlike other molecules, spherical top molecules have no net dipole moment, and, hence, they do not exhibit a pure rotational spectrum.
Asymmetric top
The spectrum for these molecules usually involves many lines due to three different rotational modes and their combinations. The following analysis is valid for the general case and collapses to the various special cases described above in the appropriate limit.
From the moments of inertia one can define an asymmetry parameter κ as
 , which varies from -1 for a prolate symmetric top to 1 for an oblate symmetric top.
, which varies from -1 for a prolate symmetric top to 1 for an oblate symmetric top.
One can define a scaled rotational Hamiltonian dependent on J and κ. The (symmetric) matrix representation of this Hamiltonian is banded, zero everywhere but the main diagonal and the second subdiagonal. The Hamiltonian can be formulated in six different settings, dependent on the mapping of the principal axes to lab axes and handedness. For the most asymmetric, right-handed representation, the diagonal elements are, for

- Hk,k(κ) = κk2
and the second off-diagonal elements (independent of κ) are
![H_{k,k+2}(\kappa)=\sqrt{[J(J+1)-(k+1)(k+2)][J(J+1)-k(k+1)]}/2](4/6a4b6ec66ef530bfe969877601a14cf7.png) .
.
Diagonalising H yields a set of 2J + 1 scaled rotational energy levels Ek(κ). The rotational energy levels of the asymmetric rotor for total angular momentum J are then given by
Hyperfine interaction
In addition to the main structure that is observed in microwave spectra due to the rotational motion of the molecules, a whole host of further interactions are responsible for small details in the spectra, and the study of these details provides a very deep understanding of molecular quantum mechanics. The main interactions responsible for small changes in the spectra (additional splittings and shifts of lines) are due to magnetic and electrostatic interactions in the molecule. The particular strength of such interactions differs in different molecules, but, in general, the order of these effects (in decreasing significance) is:
- electron spin - electron spin interaction (this occurs in molecules with two or more unpaired electrons, and is a magnetic-dipole / magnetic-dipole interaction)
- electron spin - molecular rotation (the rotation of a molecule corresponds to a magnetic dipole, which interacts with the magnetic dipole moment of the electron)
- electron spin - nuclear spin interaction (the interaction between the magnetic dipole moment of the electron and the magnetic dipole moment of the nuclei (if present)).
- electric field gradient - nuclear electric quadrupole interaction (the interaction between the electric field gradient of the electron cloud of the molecule and the electric quadrupole moments of nuclei (if present)).
- nuclear spin - nuclear spin interaction (nuclear magnetic moments interacting with one another).
These interactions give rise to the characteristic energy levels that are probed in "magnetic resonance" spectroscopy such as NMR and ESR, where they represent the "zero field splittings," which are always present.
Experimental determination of the spectrum
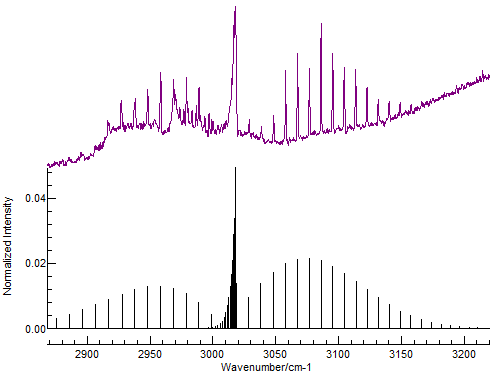
See also: Rovibrational coupling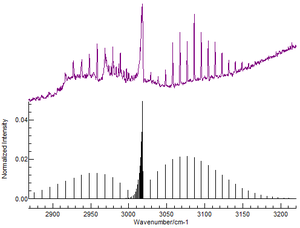 Part of the rotational-vibrational spectrum of methane (CH4) gas (from FTIR), showing the presence of P-, Q- and R- branches (purple, top) and a simulation in PGOPHER (black, bottom). Frequency is on the x-axis, and transmittance on the y-axis.
Part of the rotational-vibrational spectrum of methane (CH4) gas (from FTIR), showing the presence of P-, Q- and R- branches (purple, top) and a simulation in PGOPHER (black, bottom). Frequency is on the x-axis, and transmittance on the y-axis.Fourier transform infrared (FTIR) spectroscopy can be used to experimentally study rotational spectra. Typical spectra at these wavelengths involve rovibrational excitation, i.e., excitation of both a vibrational and a rotational mode of a molecule.
In the past, microwave spectra were determined using a simple arrangement in which low-pressure gas was introduced to a section of waveguide between a microwave source (of variable frequency) and a microwave detector. The spectrum was obtained by sweeping the frequency of the source while detecting the intensity of the transmitted radiation. This experimental arrangement has a major difficulty related to the propagation of microwave radiation through waveguides. The physical size of the waveguide restricts the frequency of the radiation that can be transmitted through it. For a given waveguide size (such as X-band), there is a cutoff frequency, and microwave radiation with smaller frequencies (longer wavelengths) cannot be propagated through the waveguide. In addition, as the frequency is increased, additional modes of propagation become possible, which correspond to different velocities of the radiation propagating down the waveguide (this can be envisaged as the radiation bouncing down the guide, at different angles of reflection). the net result of these considerations is that each size of waveguide is useful only over a rather narrow range of frequencies and must be physically swapped out for a different size of waveguide once this frequency range is exceeded.
From 1980 onward, microwave spectra have often been obtained using Fourier Transform Microwave Spectroscopy - a technique developed by W. H. Flygare at the University of Illinois.
Within the last two years, a further development of Fourier Transform Microwave Spectroscopy has occurred, which may well introduce a new renaissance into microwave spectroscopy. This is the use of "Chirped Pulses" to provide an electromagnetic wave that has as its Fourier Transform a very wide range of microwave frequencies. (see University of Virginia, Chemistry Department website).
Applications
Microwave spectroscopy is commonly used in physical chemistry to determine the structure of small molecules (such as ozone, methanol, or water) with high precision. Other common techniques for determining molecular structure, such as X-ray crystallography do not work very well for some of these molecules (especially the gases) and are not as precise. However, microwave spectroscopy is not useful for determining the structures of large molecules such as proteins.
Modern microwave spectrometers have very high resolution. When hyperfine structure can be observed, the technique can also provide information on the electronic structures of molecules.
Microwave spectroscopy is one of the principal means by which the constituents of the universe are determined from the earth. It is particularly useful for detecting molecules in the interstellar medium (ISM). One of the early surprises in interstellar chemistry came with the discovery of the existence in the ISM of long-chain carbon molecules. It was in attempting to research such molecules in the laboratory that Harry Kroto was led to the laboratory of Rick Smalley and Robert Curl, where it was possible to vaporize carbon under enormous energy conditions. This collaborative experiment led to the discovery of C60, buckminsterfullerene, which led to the award of the 1996 Nobel prize in chemistry to Kroto, Smalley and Curl.
See also
- Spectroscopy
- Rigid rotor
- Rovibronic excitation
- Vibrational spectroscopy
- Infrared spectroscopy
References
- Microwave Spectroscopy, Townes and Schawlow, Dover;
- Molecular Rotation Spectra, Harry Kroto, Dover;
- Rotational Spectroscopy of Diatomic molecules, Brown and Carrington;
- Quantum Mechanics, Mcquarrie, Donald A.
External links
- infrared gas spectra simulator
- Hyperphysics article on Rotational Spectrum
- A list of microwave spectroscopy research groups around the world
Categories:












Wikimedia Foundation. 2010.