- Systemic primary carnitine deficiency
-
- CUD is an acronym sometimes used for primary carnitine deficiency. For other uses, see Cud (disambiguation).
Systemic primary carnitine deficiency Classification and external resources 
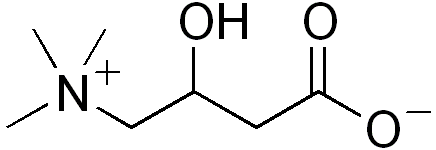
CarnitineICD-10 E71.3 ICD-9 277.81 OMIM 212140 DiseasesDB 31103 eMedicine ped/321 Systemic primary carnitine deficiency (CDSP), also called deficiency of plasma-membrane carnitine transporter, carnitine transporter deficiency (CTD) or carnitine uptake defect (CUD),[1] is an autosomal recessive[2] metabolic disorder that prevents the body from using fats for energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through diet,[3] is used by cells to process fats and produce energy. People with primary carnitine deficiency have defective proteins called carnitine transporters, which bring carnitine into cells and prevent its escape from the body.
Contents
Signs and symptoms
Typically, signs and symptoms of this disorder appear during infancy or early childhood and often include brain function abnormalities (encephalopathy); an enlarged, poorly pumping heart (cardiomyopathy); confusion; vomiting; muscle weakness; and low blood sugar (hypoglycemia). Serious complications such as heart failure, liver problems, coma, and sudden unexpected death are also a risk. Acute illness due to primary carnitine deficiency can be triggered by periods of fasting or illnesses such as viral infections, particularly when eating is reduced.
Cause and genetics
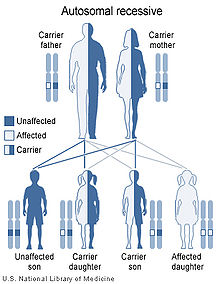
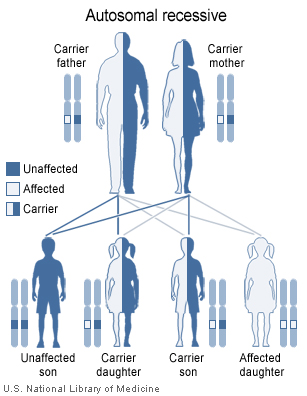
CDSP is caused by mutations in the SLC22A5 gene, located on chromosome 5q31.1.[4] Improper function of this gene leads to the production of defective carnitine transporters. As a result of reduced transport function, carnitine is lost from the body and cells are not supplied with an adequate amount of carnitine. Without carnitine, fats cannot be processed correctly and are not converted into energy, which can lead to characteristic signs and symptoms of this disorder.
It is an autosomal recessive disorder,[1] which means the defective gene responsible for the disorder is located on an autosome (chromosome 5 is an autosome), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Diagnosis and treatment
CDSP is sometimes diagnosed in adults and is then thought to be less severe both in symptoms and life expectation. Treatment is usually done by supplementation of L-carnitine after assessing the severity of the deficiency after a muscular biopsy.
This condition is sometimes mistaken for Reye's syndrome, a severe disorder that develops in children while they appear to be recovering from viral infections such as chicken pox or flu. Most cases of Reye syndrome are associated with the use of aspirin during these viral infections.
Epidemiology
On the average, primary carnitine deficiency affects 1 in every 40,000 live births in Japan and 1 in every 37,000 to 100,000 newborns in Australia. Of particular interest is the population of the Faroe Islands, in which the condition is found in about 1 in every 500 people.[5] The incidence of this condition in other populations has not been determined.[citation needed]
History
The current understanding of primary carnitine deficiency has been greatly influenced by the research of Doctors Susan C. Winter and Neil Buist. Dr. Winter was one of the first doctors in the United States to begin treating inborn errors of metabolism with intravenous carnitine.[citation needed]
References
- ^ a b Online 'Mendelian Inheritance in Man' (OMIM) 212140
- ^ El-Hattab, A. W.; Li, F. Y.; Shen, J.; Powell, B. R.; Bawle, E. V.; Adams, D. J.; Wahl, E.; Kobori, J. A. et al. (Jan 2010). "Maternal systemic primary carnitine deficiency uncovered by newborn screening: Clinical, biochemical, and molecular aspects". Genetics in Medicine 12 (1): 19–24. doi:10.1097/GIM.0b013e3181c5e6f7. PMID 20027113.
- ^ Primary carnitine deficiency from Genetics Home Reference, a service of the U.S. National Library of Medicine. Reviewed July 2006
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 603377
- ^ "Faeroes susceptible to deadly illness", The Copenhagen Post (Copenhagen), 17 May 2010, http://www.cphpost.dk/news/135-science/48995-faeroes-susceptible-to-deadly-illness.html
External links
This article incorporates public domain text from The U.S. National Library of Medicine
Inborn error of lipid metabolism: fatty-acid metabolism disorders (E71.3, 277.81–277.85) Synthesis Degradation Acyl transportGeneralOtherTo acetyl-CoASjögren–Larsson syndromeCategories:- Autosomal recessive disorders
- Hepatology
- Fatty-acid metabolism disorders
Wikimedia Foundation. 2010.