- Autophagy
-
This article is about the cellular process. For other uses, see Autophagy (disambiguation).
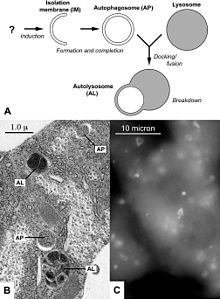 (A) Diagram of autophagy; (B) Electron micrograph of autophagic structures in the fatbody of a fruit fly larva; (C) Fluorescently labeled autophagosomes in liver cells of starved mice.
(A) Diagram of autophagy; (B) Electron micrograph of autophagic structures in the fatbody of a fruit fly larva; (C) Fluorescently labeled autophagosomes in liver cells of starved mice.
In cell biology, autophagy, or autophagocytosis, is a catabolic process involving the degradation of a cell's own components through the lysosomal machinery. It is a tightly regulated process that plays a normal part in cell growth, development, and homeostasis, helping to maintain a balance between the synthesis, degradation, and subsequent recycling of cellular products. It is a major mechanism by which a starving cell reallocates nutrients from unnecessary processes to more-essential processes.
A variety of autophagic processes exist, all having in common the degradation of intracellular components via the lysosome. The most well-known mechanism of autophagy involves the formation of a membrane around a targeted region of the cell, separating the contents from the rest of the cytoplasm. The resultant vesicle then fuses with a lysosome and subsequently degrades the contents.
It was first described in the 1960s,[1][2] but many questions about the actual processes and mechanisms involved still remain to be elucidated. Its role in disease is not well categorized; it may help to prevent or halt the progression of some diseases such as some types of neurodegeneration and cancer,[3] and play a protective role against infection by intracellular pathogens; however, in some situations, it may actually contribute to the development of a disease.[4]
Contents
Etymology
Autophagy is derived from Greek roots: auto, meaning 'self', and phagy, 'to eat'.
Selective autophagy
- Pexophagy, autophagy selective for degradation of peroxisomes, which can be separated into macropexophagy and micropexophagy.[5]
- Mitophagy, autophagy selective for degradation of mitochondria, which can be separated into macromitophagy and micromitophagy.
- Xenophagy, autophagy selective for degradation of intracellular bacteria and viruses (foreign bodies).
- Aggrephagy, autophagy selective for protein aggregates.
- Reticulophagy, autophagy selective for endoplasmic reticulum.
- Heterophagy, autophagy selective for endosomes.
- Crinophagy, autophagy selective for golgi apparatus.
- Ribophagy, autophagy selective for ribosomes.[6]
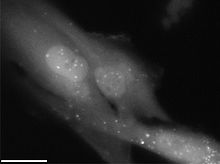 Autophagosomes labeled by a fluorescent marker.
Autophagosomes labeled by a fluorescent marker.Process
Macroautophagy sequestrates damaged organelles and unused long-lived proteins in a double-membrane vesicle, called an autophagosome or autophagic vacuole (AV), inside the cell. Autophagosomes form from the elongation of small membrane structures known as autophagosome precursors. The formation of autophagosomes is initiated by class III phosphoinositide 3-kinase and autophagy-related gene (Atg) 6 (also known as Beclin-1). In addition, two further systems are involved, composed of the ubiquitin-like protein Atg8 (known as LC3 in mammalian cells) and the Atg4 protease on the one hand and the Atg12-Atg5-Atg16 complex on the other.[7] The outer membrane of the autophagosome fuses in the cytoplasm with a lysosome to form an autolysosome or autophagolysosome where their contents are degraded via acidic lysosomal hydrolases.[8]
Microautophagy, on the other hand, happens when lysosomes directly engulf cytoplasm by invaginating, protrusion, and/or septation of the lysosomal limiting membrane.
In Chaperone-mediated autophagy, or CMA, only those proteins that have a consensus peptide sequence get recognized by the binding of a hsc70-containing chaperone/co-chaperone complex. This CMA substrate/chaperone complex then moves to the lysosomes, where the CMA receptor lysosome-associated membrane protein type-2A (LAMP-2A) recognizes it; the protein is unfolded and translocated across the lysosome membrane assisted by the lysosomal hsc70 on the other side. CMA differs from macroautophagy and microautophagy in two main ways:
- The substrates are translocated across the lysosome membrane on a one-by-one basis, whereas in the macroautophagy and microautophagy the substrates are engulfed or sequestered in-bulk.
- CMA is very selective in what it degrades and can degrade only certain proteins and not organelles.
Autophagy is part of everyday normal cell growth and development wherein mTOR plays an important regulatory role.
Functions
Nutrient starvation
During nutrient starvation, increased levels of autophagy lead to the breakdown of non-vital components and the release of nutrients, ensuring that vital processes can continue.[9] Mutant yeast cells that have a reduced autophagic capability rapidly perish in nutrition-deficient conditions.[10] A gene known as Atg7 has been implicated in nutrient-mediated autophagy, as mice studies have shown that starvation-induced autophagy was impaired in Atg7-deficient mice.[11]
Infection
Autophagy plays a role in the destruction of some bacteria within the cell. Intracellular pathogens such as Mycobacterium tuberculosis persist within cells and block the normal actions taken by the cell to rid itself of it. Stimulating autophagy in infected cells overcomes the block and helps to rid the cell of pathogens.[12] In addition to "simple" breakdown of pathogens, it has also been shown that at least in some cell types (plasmacytoid dendritic cells) autophagy play a role in detection of virus by the so-called pattern recognition receptors (PRR), which are part of the innate immune system.[13] The virus (Vesicular stomatitis virus) is believed to be taken up by the autophagosome from the cytosol and translocated to the endosomes where detection takes place by a member of the PRRs called toll-like receptor 7, detecting single-stranded RNA. Following activation of the toll-like receptor, intracellular signalling cascades are initiated, leading to induction of interferon, among other anti-viral cytokines. A subset of viruses and bacteria subvert the autophagic pathway to promote their own replication. (http://www.plosbiology.org/article/info:doi/10.1371/journal.pbio.0030156)
Repair mechanism
Autophagy degrades damaged organelles, cell membranes and proteins, and the failure of autophagy is thought to be one of the main reasons for the accumulation of cell damage and aging.
Programmed cell death
It has been proposed that autophagy resulting in the total destruction of the cell is one of several types of programmed cell death; yet, no conclusive evidence exists for such a process.[14] Nevertheless, observations that cells possessing autophagic features in areas undergoing programmed cell death have led to the coining of the phrase autophagic cell death (also known as cytoplasmic cell death or type II cell death). Studies of the metamorphosis of insects have shown cells undergoing a form of programmed cell death that appears distinct from other forms; these have been proposed as examples of autophagic cell death.[15]
It is not known whether autophagic activity in dying cells actually causes cell death or whether it simply occurs as a process alongside it. In many neurological diseases, in certain neuronal cell death pathways and after neuronal injury, there are increased numbers of autophagosomes. A causative relationship between autophagy and cell death has not been established. It is unclear whether the increase in autophagosomes indicates an increase in autophagic activity or decreased autophagosome-lysosome fusion.[8] Recently it has been argued that autophagy might actually be a survival mechanism on behalf of the cell.[14]
Examples
Autophagia can occur in body cells as a method of sustaining the life of a cell. Alternatively, the term could apply to an organism recycling tissue for sustenance. In myeloid precursor cells, autophagia can be an indicator of CHS, and a possible explanation for neutropenia.[16]
Certain diets utilize a form of autophagia. The Atkins Diet relies heavily on ketosis as a method of reducing body fat, which, in itself, could be considered a form of cellular autophagia.[citation needed]
Academic research concerning oncological pharmacology and autophagy
According to a Wednesday, November 9, 2011 online news article (the story was released by David Cameron of FOCUS, which publishes news from Harvard Medical School, Harvard School of Dental Medicine, and the Harvard School of Public Health):
"Researchers have discovered a small molecule that disables a prized cancer target, one that many pharmaceutical and biotech companies have been investigating for years.
The findings, which also establish a chain linking the target to the tumor-suppressing gene p53, suggest a long-sought weapon against the defenses of cancer cells.
The results were published in the journal "Cell".
The process, called autophagy, rids cells of debris and is crucial for cell survival.
“Autophagy helps cells survive stress,” said Junying Yuan, Harvard Medical School professor of cell biology and senior author on the paper. “It’s like a recycling process that degrades old proteins into amino acid energy sources enabling cells to survive in difficult circumstances. It’s a turnover mechanism.”
When autophagy falters, life span shortens, and cancer and other diseases, such as neurodegeneration, can ensue. One such defect, in a gene called Beclin1, decreases autophagy in mammalian cells, and researchers have suspected that this leads to increased prostate and breast cancers.
But like so many cancer factors, autophagy can be a double-edged sword.
When a patient is undergoing treatment such as chemotherapy, cancer cells co-opt autophagy and use it to survive the stress of therapy. Researchers have reasoned that in certain clinical settings, briefly disabling autophagy may support and enhance treatment.
For years, pharmaceutical companies have sought to do just that. The challenge lay in identifying the precise target within a protein complex. Yuan and her colleagues developed a cell-based screening platform in which they uncovered a key mechanism of autophagy as well as a small molecule that efficiently blocks the process by degrading the protein complex that autophagy depends on. The protein beclin1, encoded by the Beclin1 gene already linked to autophagy, is a part of this complex.
They named the molecule spautin-1, for specific and potent autophagy inhibitor-1.
Drilling deeper, the researchers found that spautin-1 blocked the activity of USP10, a molecule that offers a kind of “stay of execution” for proteins on death row. Proteins marked for disposal are tagged with a marker called ubiquitin, and USP10 often removes this tag from select proteins, sparing them. Removing USP10 leaves these proteins vulnerable.
Beclin1, it turns out, regulates the activity of USP10. And the researchers connected these findings to other studies linking USP10 to p53, a gene widely known to suppress cancer.
“Knocking down Beclin1, which our small molecule does, knocks down USP10, which in turn knocks down p53,” said Yuan. “They are all part of a chain.”
This then explains the earlier observation that mammals with defective Beclin1 experience increased cancer. When beclin1 is diminished, p53, which is downstream, is also diminished, and cancer thrives. However, when Beclin1 is removed altogether, the cell dies. This discovery suggests that selectively targeting autophagy during cancer therapies may greatly benefit patients.
Yuan is now collaborating with researchers at the company Roche, based in Basel, Switzerland and at BioBay, based in Suzhou, China, to translate these findings into potential therapies.
This research was funded by the National Institutes of Health, the Chinese Academy of Sciences, the National Natural Science Foundation of China, and the Harvard University Biomedical Accelerator Fund.
—David Cameron One Response to Long Sought Cancer"[17]
See also
- Autophagy database
- Autophagin
- Apoptosis
- Sub-lethal damage
- Ubiquitin
External links
- Autophagy, a journal produced by Landes Bioscience and edited by DJ Klionsky
- LongevityMeme entry describing PubMed article on the effects of autophagy and lifespan
- Autophagolysosome on Drugs.com
- HADb, a Human Autophagy dedicated Database
- Autophagy DB, an autophagy database that covers all eukaryotes
- Self-Destructive Behavior in Cells May Hold Key to a Longer Life
References
- ^ Stromhaug PE, Klionsky DJ (2001). "Approaching the Molecular Mechanism of Autophagy". Traffic 2 (8): 524–531. doi:10.1034/j.1600-0854.2001.20802.x. PMID 11489210.
- ^ Kundu M, Thompson CB (2008). "Autophagy: Basic Principles and Relevance to Disease". Annual Review of Pathology 3: 427–455. doi:10.1146/annurev.pathmechdis.2.010506.091842. PMID 18039129. http://arjournals.annualreviews.org/doi/full/10.1146/annurev.pathmechdis.2.010506.091842.
- ^ Mizushima N, Klionsky DJ (2007). "Protein Turnover Via Autophagy: Implications for Metabolism". Annual Review of Nutrition 27: 19–40. doi:10.1146/annurev.nutr.27.061406.093749. PMID 17311494. http://arjournals.annualreviews.org/doi/abs/10.1146/annurev.nutr.27.061406.093749.
- ^ Ling, Daijun; Song, Ho-Juhn; Garza, Dan; Neufeld, Thomas P.; Salvaterra, Paul M. (2009). Cookson, Mark R.. ed. "Abeta42-Induced Neurodegeneration via an Age-Dependent Autophagic-Lysosomal Injury in Drosophila". PLoS ONE 4 (1): e4201. doi:10.1371/journal.pone.0004201. PMC 2626277. PMID 19145255. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2626277.
- ^ Goldman, Scott J; Robert Taylor, Yong Zhang, Shengkan Jin (2010-01-18). "Autophagy and the degradation of mitochondria". Mitochondrion 10 (4): 309–15. doi:10.1016/j.mito.2010.01.005. ISSN 1872-8278. PMC 2874649. PMID 20083234. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2874649.
- ^ Peter, Kraft C; Anna Deplazes, Marc Sohrmann (2008-04-06). "Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease". Nature Cell Biology 10 (5): 602–610. doi:10.1038/ncb1723. PMID 18391941.
- ^ Schmid, D; Münz, C (2007). "Innate and adaptive immunity through autophagy.". Immunity 27 (1): 11–21. doi:10.1016/j.immuni.2007.07.004. PMID 17663981.
- ^ a b Rubinsztein DC et al. (2005). "Autophagy and Its Possible Roles in Nervous System Diseases, Damage and Repair". Autophagy 1 (1): 11–22. PMID 16874045.
- ^ Yorimitsu T, Klionsky DJ (2005). "Autophagy: molecular machinery for self-eating". Cell Death and Differentiation (2005) 12, 1542–1552 12: 1542–1552. doi:10.1038/sj.cdd.4401765. PMC 1828868. PMID 16247502. http://www.nature.com/cdd/journal/v12/n2s/full/4401765a.html.
- ^ Tsukada M, Ohsumi Y (1993). "Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae". FEBS Lett. 333 (1-2): 169–174. doi:10.1016/0014-5793(93)80398-E. PMID 8224160.
- ^ M Komatsu et al. (2005). "Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice". JCB 169 (3): 425–434. doi:10.1083/jcb.200412022. PMC 2171928. PMID 15866887. http://www.jcb.org/cgi/content/full/169/3/425#BIB41.
- ^ Gutierrez MG et al. (2004). "Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages". Cell 119 (6): 753–766. doi:10.1016/j.cell.2004.11.038. PMID 15607973.
- ^ http://www.sciencemag.org/cgi/content/abstract/sci;315/5817/1398
- ^ a b Tsujimoto Y, Shimizu S (2005). "Another way to die: autophagic programmed cell death". Cell Death and Differentiation 12: 1528–1534. doi:10.1038/sj.cdd.4401777. PMID 16247500. http://www.nature.com/cdd/journal/v12/n2s/full/4401777a.html.
- ^ Schwartz LM, et al. (1993). "Do All Programmed Cell Deaths Occur Via Apoptosis?". Proceedings of the National Academy of Sciences 90 (3): 980–984. doi:10.1073/pnas.90.3.980. PMC 45794. PMID 8430112. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=45794.
- ^ Oberling F, Lang JM, Juif JG, Luckel JC (August 1976). "Autophagia in myeloid precursors: an explanation for neutropenia in Chediak-Higashi syndrome?". Scand J Haematol 17 (2): 105–10. doi:10.1111/j.1600-0609.1976.tb01162.x. PMID 968442.
- ^ http://www.focushms.com/features/long-sought-cancer-target-unraveled/
Structures of the cell / organelles (TH H1.00.01.2-3) Endomembrane system Cytoskeleton Endosymbionts Other internal External Categories:- Cellular processes
- Programmed cell death


Wikimedia Foundation. 2010.