- Sequence alignment
-
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences.[1] Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns.
 A sequence alignment, produced by ClustalW, of two human zinc finger proteins, identified on the left by GenBank accession number.
A sequence alignment, produced by ClustalW, of two human zinc finger proteins, identified on the left by GenBank accession number.
Key: Single letters: amino acids. Red: small, hydrophobic, aromatic, not Y. Blue: acidic. Magenta: basic. Green: hydroxyl, amine, amide, basic. Gray: others. "*": identical. ":": conserved substitutions (same colour group). ".": semi-conserved substitution (similar shapes).[2]Sequence alignments are also used for non-biological sequences, such as those present in natural language or in financial data.
Contents
Interpretation
If two sequences in an alignment share a common ancestor, mismatches can be interpreted as point mutations and gaps as indels (that is, insertion or deletion mutations) introduced in one or both lineages in the time since they diverged from one another. In sequence alignments of proteins, the degree of similarity between amino acids occupying a particular position in the sequence can be interpreted as a rough measure of how conserved a particular region or sequence motif is among lineages. The absence of substitutions, or the presence of only very conservative substitutions (that is, the substitution of amino acids whose side chains have similar biochemical properties) in a particular region of the sequence, suggest [3] that this region has structural or functional importance. Although DNA and RNA nucleotide bases are more similar to each other than are amino acids, the conservation of base pairs can indicate a similar functional or structural role.
Alignment methods
Very short or very similar sequences can be aligned by hand. However, most interesting problems require the alignment of lengthy, highly variable or extremely numerous sequences that cannot be aligned solely by human effort. Instead, human knowledge is applied in constructing algorithms to produce high-quality sequence alignments, and occasionally in adjusting the final results to reflect patterns that are difficult to represent algorithmically (especially in the case of nucleotide sequences). Computational approaches to sequence alignment generally fall into two categories: global alignments and local alignments. Calculating a global alignment is a form of global optimization that "forces" the alignment to span the entire length of all query sequences. By contrast, local alignments identify regions of similarity within long sequences that are often widely divergent overall. Local alignments are often preferable, but can be more difficult to calculate because of the additional challenge of identifying the regions of similarity. A variety of computational algorithms have been applied to the sequence alignment problem, including slow but formally correct methods like dynamic programming, and efficient, heuristic algorithms or probabilistic methods that do not guarantee to find best matches designed for large-scale database search.
Representations
Alignments are commonly represented both graphically and in text format. In almost all sequence alignment representations, sequences are written in rows arranged so that aligned residues appear in successive columns. In text formats, aligned columns containing identical or similar characters are indicated with a system of conservation symbols. As in the image above, an asterisk or pipe symbol is used to show identity between two columns; other less common symbols include a colon for conservative substitutions and a period for semiconservative substitutions. Many sequence visualization programs also use color to display information about the properties of the individual sequence elements; in DNA and RNA sequences, this equates to assigning each nucleotide its own color. In protein alignments, such as the one in the image above, color is often used to indicate amino acid properties to aid in judging the conservation of a given amino acid substitution. For multiple sequences the last row in each column is often the consensus sequence determined by the alignment; the consensus sequence is also often represented in graphical format with a sequence logo in which the size of each nucleotide or amino acid letter corresponds to its degree of conservation.[4]
Sequence alignments can be stored in a wide variety of text-based file formats, many of which were originally developed in conjunction with a specific alignment program or implementation. Most web-based tools allow a limited number of input and output formats, such as FASTA format and GenBank format and the output is not easily editable. Several conversion programs are available,[dead link] READSEQ or EMBOSS having a graphical interfaces or command line interfaces, while several programming packages like BioPerl, BioRuby provide functions to do this.
Global and local alignments
 Illustration of global and local alignments demonstrating the 'gappy' quality of global alignments that can occur if sequences are insufficiently similar
Illustration of global and local alignments demonstrating the 'gappy' quality of global alignments that can occur if sequences are insufficiently similarGlobal alignments, which attempt to align every residue in every sequence, are most useful when the sequences in the query set are similar and of roughly equal size. (This does not mean global alignments cannot end in gaps.) A general global alignment technique is the Needleman–Wunsch algorithm, which is based on dynamic programming. Local alignments are more useful for dissimilar sequences that are suspected to contain regions of similarity or similar sequence motifs within their larger sequence context. The Smith–Waterman algorithm is a general local alignment method also based on dynamic programming. With sufficiently similar sequences, there is no difference between local and global alignments.
Hybrid methods, known as semiglobal or "glocal" (short for global-local) methods, attempt to find the best possible alignment that includes the start and end of one or the other sequence. This can be especially useful when the downstream part of one sequence overlaps with the upstream part of the other sequence. In this case, neither global nor local alignment is entirely appropriate: a global alignment would attempt to force the alignment to extend beyond the region of overlap, while a local alignment might not fully cover the region of overlap.[5]
Pairwise alignment
Pairwise sequence alignment methods are used to find the best-matching piecewise (local) or global alignments of two query sequences. Pairwise alignments can only be used between two sequences at a time, but they are efficient to calculate and are often used for methods that do not require extreme precision (such as searching a database for sequences with high similarity to a query). The three primary methods of producing pairwise alignments are dot-matrix methods, dynamic programming, and word methods;[1] however, multiple sequence alignment techniques can also align pairs of sequences. Although each method has its individual strengths and weaknesses, all three pairwise methods have difficulty with highly repetitive sequences of low information content - especially where the number of repetitions differ in the two sequences to be aligned. One way of quantifying the utility of a given pairwise alignment is the 'maximum unique match' (MUM), or the longest subsequence that occurs in both query sequence. Longer MUM sequences typically reflect closer relatedness.
Dot-matrix methods
 A DNA dot plot of a human zinc finger transcription factor (GenBank ID NM_002383), showing regional self-similarity. The main diagonal represents the sequence's alignment with itself; lines off the main diagonal represent similar or repetitive patterns within the sequence. This is a typical example of a recurrence plot.
A DNA dot plot of a human zinc finger transcription factor (GenBank ID NM_002383), showing regional self-similarity. The main diagonal represents the sequence's alignment with itself; lines off the main diagonal represent similar or repetitive patterns within the sequence. This is a typical example of a recurrence plot.
The dot-matrix approach, which implicitly produces a family of alignments for individual sequence regions, is qualitative and conceptually simple, though time-consuming to analyze on a large scale. In the absence of noise, it can be easy to visually identify certain sequence features—such as insertions, deletions, repeats, or inverted repeats—from a dot-matrix plot. To construct a dot-matrix plot, the two sequences are written along the top row and leftmost column of a two-dimensional matrix and a dot is placed at any point where the characters in the appropriate columns match—this is a typical recurrence plot. Some implementations vary the size or intensity of the dot depending on the degree of similarity of the two characters, to accommodate conservative substitutions. The dot plots of very closely related sequences will appear as a single line along the matrix's main diagonal.
Problems with dot plots as an information display technique include: noise, lack of clarity, non-intuitiveness, difficulty extracting match summary statistics and match positions on the two sequences. There is also much wasted space where the match data is inherently duplicated across the diagonal and most of the actual area of the plot is taken up by either empty space or noise, and, finally, dot-plots are limited to two sequences. None of these limitations apply to Miropeats alignment diagrams but they have their own particular flaws.
Dot plots can also be used to assess repetitiveness in a single sequence. A sequence can be plotted against itself and regions that share significant similarities will appear as lines off the main diagonal. This effect can occur when a protein consists of multiple similar structural domains.
Dynamic programming
The technique of dynamic programming can be applied to produce global alignments via the Needleman-Wunsch algorithm, and local alignments via the Smith-Waterman algorithm. In typical usage, protein alignments use a substitution matrix to assign scores to amino-acid matches or mismatches, and a gap penalty for matching an amino acid in one sequence to a gap in the other. DNA and RNA alignments may use a scoring matrix, but in practice often simply assign a positive match score, a negative mismatch score, and a negative gap penalty. (In standard dynamic programming, the score of each amino acid position is independent of the identity of its neighbors, and therefore base stacking effects are not taken into account. However, it is possible to account for such effects by modifying the algorithm.) A common extension to standard linear gap costs, is the usage of two different gap penalties for opening a gap and for extending a gap. Typically the former is much larger than the latter, e.g. -10 for gap open and -2 for gap extension. Thus, the number of gaps in an alignment is usually reduced and residues and gaps are kept together, which typically makes more biological sense. The Gotoh algorithm implements affine gap costs by using three matrices.
Dynamic programming can be useful in aligning nucleotide to protein sequences, a task complicated by the need to take into account frameshift mutations (usually insertions or deletions). The framesearch method produces a series of global or local pairwise alignments between a query nucleotide sequence and a search set of protein sequences, or vice versa. Its ability to evaluate frameshifts offset by an arbitrary number of nucleotides makes the method useful for sequences containing large numbers of indels, which can be very difficult to align with more efficient heuristic methods. In practice, the method requires large amounts of computing power or a system whose architecture is specialized for dynamic programming. The BLAST and EMBOSS suites provide basic tools for creating translated alignments (though some of these approaches take advantage of side-effects of sequence searching capabilities of the tools). More general methods are available from both commercial sources, such as FrameSearch, distributed as part of the Accelrys GCG package, and Open Source software such as Genewise.
The dynamic programming method is guaranteed to find an optimal alignment given a particular scoring function; however, identifying a good scoring function is often an empirical rather than a theoretical matter. Although dynamic programming is extensible to more than two sequences, it is prohibitively slow for large numbers of or extremely long sequences.
Word methods
Word methods, also known as k-tuple methods, are heuristic methods that are not guaranteed to find an optimal alignment solution, but are significantly more efficient than dynamic programming. These methods are especially useful in large-scale database searches where it is understood that a large proportion of the candidate sequences will have essentially no significant match with the query sequence. Word methods are best known for their implementation in the database search tools FASTA and the BLAST family.[1] Word methods identify a series of short, nonoverlapping subsequences ("words") in the query sequence that are then matched to candidate database sequences. The relative positions of the word in the two sequences being compared are subtracted to obtain an offset; this will indicate a region of alignment if multiple distinct words produce the same offset. Only if this region is detected do these methods apply more sensitive alignment criteria; thus, many unnecessary comparisons with sequences of no appreciable similarity are eliminated.
In the FASTA method, the user defines a value k to use as the word length with which to search the database. The method is slower but more sensitive at lower values of k, which are also preferred for searches involving a very short query sequence. The BLAST family of search methods provides a number of algorithms optimized for particular types of queries, such as searching for distantly related sequence matches. BLAST was developed to provide a faster alternative to FASTA without sacrificing much accuracy; like FASTA, BLAST uses a word search of length k, but evaluates only the most significant word matches, rather than every word match as does FASTA. Most BLAST implementations use a fixed default word length that is optimized for the query and database type, and that is changed only under special circumstances, such as when searching with repetitive or very short query sequences. Implementations can be found via a number of web portals, such as EMBL FASTA and NCBI BLAST.
Multiple sequence alignment
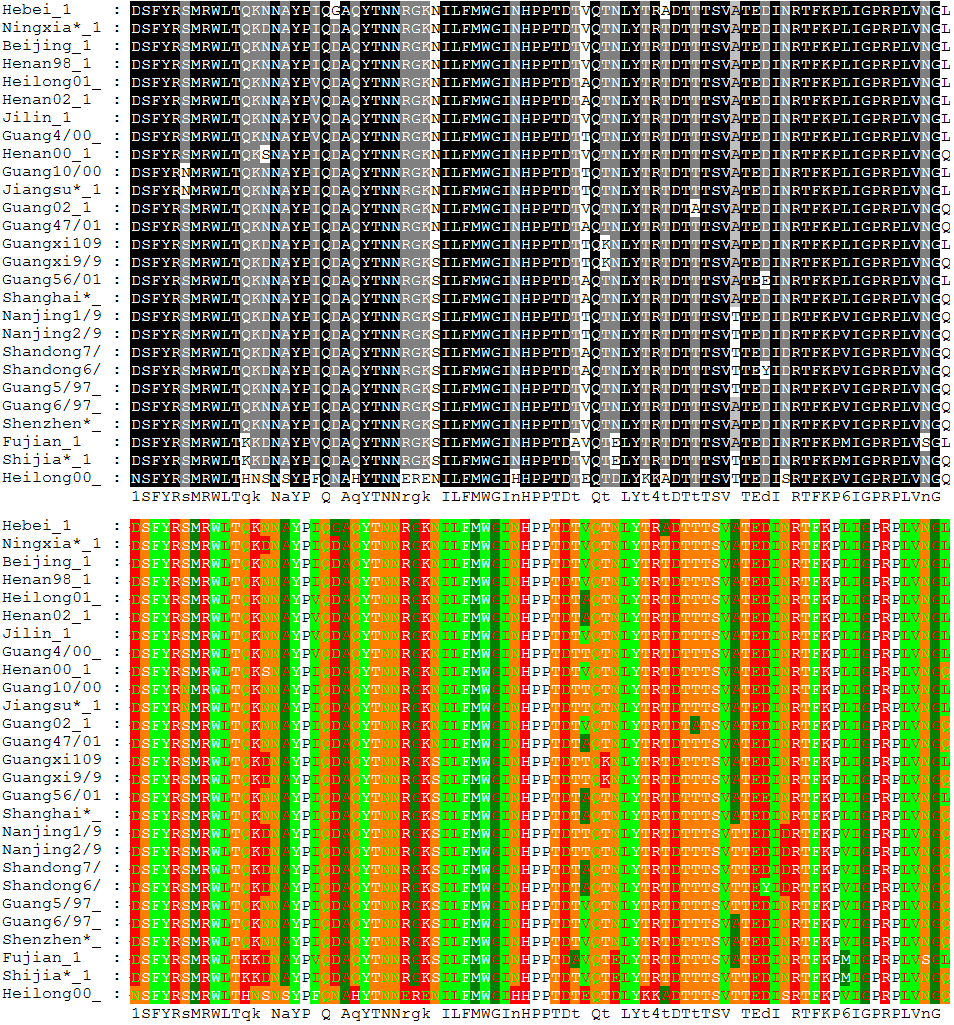
Main article: Multiple sequence alignment Alignment of 27 avian influenza hemagglutinin protein sequences colored by residue conservation (top) and residue properties (bottom)
Alignment of 27 avian influenza hemagglutinin protein sequences colored by residue conservation (top) and residue properties (bottom)Multiple sequence alignment is an extension of pairwise alignment to incorporate more than two sequences at a time. Multiple alignment methods try to align all of the sequences in a given query set. Multiple alignments are often used in identifying conserved sequence regions across a group of sequences hypothesized to be evolutionarily related. Such conserved sequence motifs can be used in conjunction with structural and mechanistic information to locate the catalytic active sites of enzymes. Alignments are also used to aid in establishing evolutionary relationships by constructing phylogenetic trees. Multiple sequence alignments are computationally difficult to produce and most formulations of the problem lead to NP-complete combinatorial optimization problems.[6][7] Nevertheless, the utility of these alignments in bioinformatics has led to the development of a variety of methods suitable for aligning three or more sequences.
Dynamic programming
The technique of dynamic programming is theoretically applicable to any number of sequences; however, because it is computationally expensive in both time and memory, it is rarely used for more than three or four sequences in its most basic form. This method requires constructing the n-dimensional equivalent of the sequence matrix formed from two sequences, where n is the number of sequences in the query. Standard dynamic programming is first used on all pairs of query sequences and then the "alignment space" is filled in by considering possible matches or gaps at intermediate positions, eventually constructing an alignment essentially between each two-sequence alignment. Although this technique is computationally expensive, its guarantee of a global optimum solution is useful in cases where only a few sequences need to be aligned accurately. One method for reducing the computational demands of dynamic programming, which relies on the "sum of pairs" objective function, has been implemented in the MSA software package.[8]
Progressive methods
Progressive, hierarchical, or tree methods generate a multiple sequence alignment by first aligning the most similar sequences and then adding successively less related sequences or groups to the alignment until the entire query set has been incorporated into the solution. The initial tree describing the sequence relatedness is based on pairwise comparisons that may include heuristic pairwise alignment methods similar to FASTA. Progressive alignment results are dependent on the choice of "most related" sequences and thus can be sensitive to inaccuracies in the initial pairwise alignments. Most progressive multiple sequence alignment methods additionally weight the sequences in the query set according to their relatedness, which reduces the likelihood of making a poor choice of initial sequences and thus improves alignment accuracy.
Many variations of the Clustal progressive implementation[9][10][11] are used for multiple sequence alignment, phylogenetic tree construction, and as input for protein structure prediction. A slower but more accurate variant of the progressive method is known as T-Coffee.[12]
Iterative methods
Iterative methods attempt to improve on the weak point of the progressive methods, the heavy dependence on the accuracy of the initial pairwise alignments. Iterative methods optimize an objective function based on a selected alignment scoring method by assigning an initial global alignment and then realigning sequence subsets. The realigned subsets are then themselves aligned to produce the next iteration's multiple sequence alignment. Various ways of selecting the sequence subgroups and objective function are reviewed in.[13]
Motif finding
Motif finding, also known as profile analysis, constructs global multiple sequence alignments that attempt to align short conserved sequence motifs among the sequences in the query set. This is usually done by first constructing a general global multiple sequence alignment, after which the highly conserved regions are isolated and used to construct a set of profile matrices. The profile matrix for each conserved region is arranged like a scoring matrix but its frequency counts for each amino acid or nucleotide at each position are derived from the conserved region's character distribution rather than from a more general empirical distribution. The profile matrices are then used to search other sequences for occurrences of the motif they characterize. In cases where the original data set contained a small number of sequences, or only highly related sequences, pseudocounts are added to normalize the character distributions represented in the motif.
Techniques inspired by computer science
A variety of general optimization algorithms commonly used in computer science have also been applied to the multiple sequence alignment problem. Hidden Markov models have been used to produce probability scores for a family of possible multiple sequence alignments for a given query set; although early HMM-based methods produced underwhelming performance, later applications have found them especially effective in detecting remotely related sequences because they are less susceptible to noise created by conservative or semiconservative substitutions.[14] Genetic algorithms and simulated annealing have also been used in optimizing multiple sequence alignment scores as judged by a scoring function like the sum-of-pairs method. More complete details and software packages can be found in the main article multiple sequence alignment.
Structural alignment
Main article: Structural alignmentStructural alignments, which are usually specific to protein and sometimes RNA sequences, use information about the secondary and tertiary structure of the protein or RNA molecule to aid in aligning the sequences. These methods can be used for two or more sequences and typically produce local alignments; however, because they depend on the availability of structural information, they can only be used for sequences whose corresponding structures are known (usually through X-ray crystallography or NMR spectroscopy). Because both protein and RNA structure is more evolutionarily conserved than sequence,[15] structural alignments can be more reliable between sequences that are very distantly related and that have diverged so extensively that sequence comparison cannot reliably detect their similarity.
Structural alignments are used as the "gold standard" in evaluating alignments for homology-based protein structure prediction[16] because they explicitly align regions of the protein sequence that are structurally similar rather than relying exclusively on sequence information. However, clearly structural alignments cannot be used in structure prediction because at least one sequence in the query set is the target to be modeled, for which the structure is not known. It has been shown that, given the structural alignment between a target and a template sequence, highly accurate models of the target protein sequence can be produced; a major stumbling block in homology-based structure prediction is the production of structurally accurate alignments given only sequence information.[16]
DALI
The DALI method, or distance matrix alignment, is a fragment-based method for constructing structural alignments based on contact similarity patterns between successive hexapeptides in the query sequences.[17] It can generate pairwise or multiple alignments and identify a query sequence's structural neighbors in the Protein Data Bank (PDB). It has been used to construct the FSSP structural alignment database (Fold classification based on Structure-Structure alignment of Proteins, or Families of Structurally Similar Proteins). A DALI webserver can be accessed at EBI DALI and the FSSP is located at The Dali Database.
SSAP
SSAP (sequential structure alignment program) is a dynamic programming-based method of structural alignment that uses atom-to-atom vectors in structure space as comparison points. It has been extended since its original description to include multiple as well as pairwise alignments,[18] and has been used in the construction of the CATH (Class, Architecture, Topology, Homology) hierarchical database classification of protein folds.[19] The CATH database can be accessed at CATH Protein Structure Classification.
Combinatorial extension
The combinatorial extension method of structural alignment generates a pairwise structural alignment by using local geometry to align short fragments of the two proteins being analyzed and then assembles these fragments into a larger alignment.[20] Based on measures such as rigid-body root mean square distance, residue distances, local secondary structure, and surrounding environmental features such as residue neighbor hydrophobicity, local alignments called "aligned fragment pairs" are generated and used to build a similarity matrix representing all possible structural alignments within predefined cutoff criteria. A path from one protein structure state to the other is then traced through the matrix by extending the growing alignment one fragment at a time. The optimal such path defines the combinatorial-extension alignment. A web-based server implementing the method and providing a database of pairwise alignments of structures in the Protein Data Bank is located at the Combinatorial Extension website.
Phylogenetic analysis
Main article: Computational phylogeneticsPhylogenetics and sequence alignment are closely related fields due to the shared necessity of evaluating sequence relatedness.[21] The field of phylogenetics makes extensive use of sequence alignments in the construction and interpretation of phylogenetic trees, which are used to classify the evolutionary relationships between homologous genes represented in the genomes of divergent species. The degree to which sequences in a query set differ is qualitatively related to the sequences' evolutionary distance from one another. Roughly speaking, high sequence identity suggests that the sequences in question have a comparatively young most recent common ancestor, while low identity suggests that the divergence is more ancient. This approximation, which reflects the "molecular clock" hypothesis that a roughly constant rate of evolutionary change can be used to extrapolate the elapsed time since two genes first diverged (that is, the coalescence time), assumes that the effects of mutation and selection are constant across sequence lineages. Therefore it does not account for possible difference among organisms or species in the rates of DNA repair or the possible functional conservation of specific regions in a sequence. (In the case of nucleotide sequences, the molecular clock hypothesis in its most basic form also discounts the difference in acceptance rates between silent mutations that do not alter the meaning of a given codon and other mutations that result in a different amino acid being incorporated into the protein.) More statistically accurate methods allow the evolutionary rate on each branch of the phylogenetic tree to vary, thus producing better estimates of coalescence times for genes.
Progressive multiple alignment techniques produce a phylogenetic tree by necessity because they incorporate sequences into the growing alignment in order of relatedness. Other techniques that assemble multiple sequence alignments and phylogenetic trees score and sort trees first and calculate a multiple sequence alignment from the highest-scoring tree. Commonly used methods of phylogenetic tree construction are mainly heuristic because the problem of selecting the optimal tree, like the problem of selecting the optimal multiple sequence alignment, is NP-hard.[22]
Assessment of significance
Sequence alignments are useful in bioinformatics for identifying sequence similarity, producing phylogenetic trees, and developing homology models of protein structures. However, the biological relevance of sequence alignments is not always clear. Alignments are often assumed to reflect a degree of evolutionary change between sequences descended from a common ancestor; however, it is formally possible that convergent evolution can occur to produce apparent similarity between proteins that are evolutionarily unrelated but perform similar functions and have similar structures.
In database searches such as BLAST, statistical methods can determine the likelihood of a particular alignment between sequences or sequence regions arising by chance given the size and composition of the database being searched. These values can vary significantly depending on the search space. In particular, the likelihood of finding a given alignment by chance increases if the database consists only of sequences from the same organism as the query sequence. Repetitive sequences in the database or query can also distort both the search results and the assessment of statistical significance; BLAST automatically filters such repetitive sequences in the query to avoid apparent hits that are statistical artifacts.
Methods of statistical significance estimation for gapped sequence alignments are available in the literature.[21][23][24][25]
Assessment of credibility
Statistical significance indicates the probability that an alignment of a given quality could arise by chance, but does not indicate how much superior a given alignment is to alternative alignments of the same sequences. Measures of alignment credibility indicate the extent to which the best scoring alignments for a given pair of sequences are substantially similar. Methods of alignment credibility estimation for gapped sequence alignments are available in the literature.[26]
Scoring functions
The choice of a scoring function that reflects biological or statistical observations about known sequences is important to producing good alignments. Protein sequences are frequently aligned using substitution matrices that reflect the probabilities of given character-to-character substitutions. A series of matrices called PAM matrices (Point Accepted Mutation matrices, originally defined by Margaret Dayhoff and sometimes referred to as "Dayhoff matrices") explicitly encode evolutionary approximations regarding the rates and probabilities of particular amino acid mutations. Another common series of scoring matrices, known as BLOSUM (Blocks Substitution Matrix), encodes empirically derived substitution probabilities. Variants of both types of matrices are used to detect sequences with differing levels of divergence, thus allowing users of BLAST or FASTA to restrict searches to more closely related matches or expand to detect more divergent sequences. Gap penalties account for the introduction of a gap - on the evolutionary model, an insertion or deletion mutation - in both nucleotide and protein sequences, and therefore the penalty values should be proportional to the expected rate of such mutations. The quality of the alignments produced therefore depends on the quality of the scoring function.
It can be very useful and instructive to try the same alignment several times with different choices for scoring matrix and/or gap penalty values and compare the results. Regions where the solution is weak or non-unique can often be identified by observing which regions of the alignment are robust to variations in alignment parameters.
Other biological uses
Sequenced RNA, such as expressed sequence tags and full-length mRNAs, can be aligned to a sequenced genome to find where there are genes and get information about alternative splicing[27] and RNA editing.[28] Sequence alignment is also a part of genome assembly, where sequences are aligned to find overlap so that contigs (long stretches of sequence) can be formed.[29] Another use is SNP analysis, where sequences from different individuals are aligned to find single basepairs that are often different in a population.[30]
Non-biological uses
The methods used for biological sequence alignment have also found applications in other fields, most notably in natural language processing and in social sciences.[31] Techniques that generate the set of elements from which words will be selected in natural-language generation algorithms have borrowed multiple sequence alignment techniques from bioinformatics to produce linguistic versions of computer-generated mathematical proofs.[32] In the field of historical and comparative linguistics, sequence alignment has been used to partially automate the comparative method by which linguists traditionally reconstruct languages.[33] Business and marketing research has also applied multiple sequence alignment techniques in analyzing series of purchases over time.[34]
Software
Main article: Sequence alignment softwareA more complete list of available software categorized by algorithm and alignment type is available at sequence alignment software, but common software tools used for general sequence alignment tasks include ClustalW and T-coffee for alignment, and BLAST and FASTA3x for database searching.
Alignment algorithms and software can be directly compared to one another using a standardized set of benchmark reference multiple sequence alignments known as BAliBASE.[35] The data set consists of structural alignments, which can be considered a standard against which purely sequence-based methods are compared. The relative performance of many common alignment methods on frequently encountered alignment problems has been tabulated and selected results published online at BAliBASE.[36] A comprehensive list of BAliBASE scores for many (currently 12) different alignment tools can be computed within the protein workbench STRAP.
References
- ^ a b c Mount DM. (2004). Bioinformatics: Sequence and Genome Analysis (2nd ed.). Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY.. ISBN 0-87969-608-7.
- ^ ClustalW2 FAQs http://www.ebi.ac.uk/Tools/clustalw2/help.html#color
- ^ Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001 May;11(5):863-74.
- ^ Schneider TD, Stephens RM (1990). "Sequence logos: a new way to display consensus sequences". Nucleic Acids Res 18 (20): 6097–6100. doi:10.1093/nar/18.20.6097. PMC 332411. PMID 2172928. http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=2172928.
- ^ Brudno M, Malde S, Poliakov A, Do CB, Couronne O, Dubchak I, Batzoglou S (2003). "Glocal alignment: finding rearrangements during alignment". Bioinformatics. 19 Suppl 1 (90001): i54–62. doi:10.1093/bioinformatics/btg1005. PMID 12855437. http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=12855437.
- ^ Wang L, Jiang T. (1994). "On the complexity of multiple sequence alignment". J Comput Biol 1 (4): 337–48. doi:10.1089/cmb.1994.1.337. PMID 8790475. http://www.liebertonline.com/doi/abs/10.1089/cmb.1994.1.337.
- ^ Elias, Isaac (2006). "Settling the intractability of multiple alignment". J Comput Biol 13 (7): 1323–1339. doi:10.1089/cmb.2006.13.1323. PMID 17037961. http://www.liebertonline.com/doi/abs/10.1089/cmb.2006.13.1323.
- ^ Lipman DJ, Altschul SF, Kececioglu JD (1989). "A tool for multiple sequence alignment". Proc Natl Acad Sci USA 86 (12): 4412–5. doi:10.1073/pnas.86.12.4412. PMC 287279. PMID 2734293. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=2734293.
- ^ Higgins DG, Sharp PM (1988). "CLUSTAL: a package for performing multiple sequence alignment on a microcomputer". Gene 73 (1): 237–44. doi:10.1016/0378-1119(88)90330-7. PMID 3243435. http://linkinghub.elsevier.com/retrieve/pii/0378-1119(88)90330-7.
- ^ Thompson JD, Higgins DG, Gibson TJ. (1994). "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice". Nucleic Acids Res 22 (22): 4673–80. doi:10.1093/nar/22.22.4673. PMC 308517. PMID 7984417. http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=7984417.[dead link]
- ^ Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. (2003). "Multiple sequence alignment with the Clustal series of programs". Nucleic Acids Res 31 (13): 3497–500. doi:10.1093/nar/gkg500. PMC 168907. PMID 12824352. http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=12824352.
- ^ Notredame C, Higgins DG, Heringa J. (2000). "T-Coffee: A novel method for fast and accurate multiple sequence alignment". J Mol Biol 302 (1): 205–17. doi:10.1006/jmbi.2000.4042. PMID 10964570. http://linkinghub.elsevier.com/retrieve/pii/S0022-2836(00)94042-7.
- ^ Hirosawa M, Totoki Y, Hoshida M, Ishikawa M. (1995). "Comprehensive study on iterative algorithms of multiple sequence alignment". Comput Appl Biosci 11 (1): 13–8. doi:10.1093/bioinformatics/11.1.13. PMID 7796270. http://bioinformatics.oxfordjournals.org/cgi/content/abstract/11/1/13.
- ^ Karplus K, Barrett C, Hughey R. (1998). "Hidden Markov models for detecting remote protein homologies". Bioinformatics 14 (10): 846–856. doi:10.1093/bioinformatics/14.10.846. PMID 9927713. http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=9927713.
- ^ Chothia C, Lesk AM. (April 1986). "The relation between the divergence of sequence and structure in proteins". EMBO J 5 (4): 823–6. PMC 1166865. PMID 3709526. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1166865.
- ^ a b Zhang Y, Skolnick J. (2005). "The protein structure prediction problem could be solved using the current PDB library". Proc Natl Acad Sci USA 102 (4): 1029–34. doi:10.1073/pnas.0407152101. PMC 545829. PMID 15653774. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=15653774.
- ^ Holm L, Sander C (1996). "Mapping the protein universe". Science 273 (5275): 595–603. doi:10.1126/science.273.5275.595. PMID 8662544. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=8662544.
- ^ Taylor WR, Flores TP, Orengo CA. (1994). "Multiple protein structure alignment". Protein Sci 3 (10): 1858–70. doi:10.1002/pro.5560031025. PMC 2142613. PMID 7849601. http://www.proteinscience.org/cgi/pmidlookup?view=long&pmid=7849601.
- ^ Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM (1997). "CATH--a hierarchic classification of protein domain structures". Structure 5 (8): 1093–108. doi:10.1016/S0969-2126(97)00260-8. PMID 9309224.
- ^ Shindyalov IN, Bourne PE. (1998). "Protein structure alignment by incremental combinatorial extension (CE) of the optimal path". Protein Eng 11 (9): 739–47. doi:10.1093/protein/11.9.739. PMID 9796821. http://peds.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=9796821.
- ^ a b Ortet P, Bastien O (2010). "Where Does the Alignment Score Distribution Shape Come from?". Evolutionary Bioinformatics 6: 159–187. doi:10.4137/EBO.S5875. PMID 21258650. http://www.la-press.com/where-does-the-alignment-score-distribution-shape-come-from-article-a2393.
- ^ Felsenstein J. (2004). Inferring Phylogenies. Sinauer Associates: Sunderland, MA. ISBN 0-87893-177-5.
- ^ Newberg LA (2008). "Significance of gapped sequence alignments". J Comput Biolo 15 (9): 1187–1194. doi:10.1089/cmb.2008.0125. PMC 2737730. PMID 18973434. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2737730.
- ^ Eddy SR; Rost, Burkhard (2008). Rost, Burkhard. ed. "A probabilistic model of local sequence alignment that simplifies statistical significance estimation". PLoS Comput Biol 4 (5): e1000069. doi:10.1371/journal.pcbi.1000069. PMC 2396288. PMID 18516236. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2396288.
- ^ Bastien O, Aude JC, Roy S, Marechal E (2004). "Fundamentals of massive automatic pairwise alignments of protein sequences: theoretical significance of Z-value statistics". Bioinformatics 20 (4): 534–537. doi:10.1093/bioinformatics/btg440. PMID 14990449. http://bioinformatics.oxfordjournals.org/content/20/4/534.long.
- ^ Newberg LA, Lawrence CE (2009). "Exact Calculation of Distributions on Integers, with Application to Sequence Alignment". J Comput Biolo 16 (1): 1–18. doi:10.1089/cmb.2008.0137. PMC 2858568. PMID 19119992. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2858568.
- ^ Kim N, Lee C (2008). "Bioinformatics detection of alternative splicing". Methods Mol. Biol. 452: 179–97. doi:10.1007/978-1-60327-159-2_9. PMID 18566765.
- ^ Li JB, Levanon EY, Yoon JK, et al. (May 2009). "Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing". Science 324 (5931): 1210–3. doi:10.1126/science.1170995. PMID 19478186.
- ^ Blazewicz J, Bryja M, Figlerowicz M, et al. (June 2009). "Whole genome assembly from 454 sequencing output via modified DNA graph concept". Comput Biol Chem 33 (3): 224–30. doi:10.1016/j.compbiolchem.2009.04.005. PMID 19477687.
- ^ Duran C, Appleby N, Vardy M, Imelfort M, Edwards D, Batley J (May 2009). "Single nucleotide polymorphism discovery in barley using autoSNPdb". Plant Biotechnol. J. 7 (4): 326–33. doi:10.1111/j.1467-7652.2009.00407.x. PMID 19386041.
- ^ Abbott A., Tsay A. (2000). "Sequence Analysis and Optimal Matching Methods in Sociology, Review and Prospect". Sociological Methods and Research 29 (1): 3–33. doi:10.1177/0049124100029001001.
- ^ Barzilay R, Lee L. (2002). "Bootstrapping Lexical Choice via Multiple-Sequence Alignment" (PDF). Proceedings of the Conference on Empirical Methods in Natural Language Processing (EMNLP) 10: 164–171. doi:10.3115/1118693.1118715. http://www.cs.cornell.edu/home/llee/papers/gen-msa.pdf.
- ^ Kondrak, Grzegorz (2002) (PDF). Algorithms for Language Reconstruction. University of Toronto, Ontario. http://www.cs.ualberta.ca/~kondrak/papers/thesis.pdf. Retrieved 2007-01-21.
- ^ Prinzie A., D. Van den Poel (2006). "Incorporating sequential information into traditional classification models by using an element/position-sensitive SAM". Decision Support Systems 42 (2): 508–526. doi:10.1016/j.dss.2005.02.004. http://econpapers.repec.org/paper/rugrugwps/05_2F292.htm. See also Prinzie and Van den Poel's paper Prinzie, A; Vandenpoel, D (2007). "Predicting home-appliance acquisition sequences: Markov/Markov for Discrimination and survival analysis for modeling sequential information in NPTB models". Decision Support Systems 44 (1): 28–45. doi:10.1016/j.dss.2007.02.008. http://econpapers.repec.org/paper/rugrugwps/07_2F442.htm.
- ^ Thompson JD, Plewniak F, Poch O (1999). "BAliBASE: a benchmark alignment database for the evaluation of multiple alignment programs". Bioinformatics 15 (1): 87–8. doi:10.1093/bioinformatics/15.1.87. PMID 10068696. http://bioinformatics.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=10068696.
- ^ Thompson JD, Plewniak F, Poch O. (1999). "A comprehensive comparison of multiple sequence alignment programs". Nucleic Acids Res 27 (13): 2682–90. doi:10.1093/nar/27.13.2682. PMC 148477. PMID 10373585. http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=10373585.
Categories:

Wikimedia Foundation. 2010.