- Mukaiyama Taxol total synthesis
-
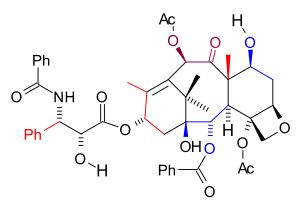 Mukaiyama Taxol total synthesis overview from raw material perspective
Mukaiyama Taxol total synthesis overview from raw material perspectiveThe Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.
This version is a linear synthesis with ring formation taking place in the order B, C, A, D. Contrary to the other published methods, the tail synthesis is by an own design. Teruaki Mukaiyama is an expert on aldol reactions and not surprisingly his Taxol version contains no less than 5 of these reactions. Other key reactions encountered in this synthesis are a pinacol coupling and a Reformatskii reaction. In terms of raw materials the C20 framework is built up from L-serine (C3), isobutyric acid (C4), glycolic acid (C2), methyl bromide (C1), methyl iodide (C1), 2,3-dibromopropene (C3), acetic acid (C2) and homoallyl bromide (C4).
Contents
Synthesis B ring
The lower rim of the cyclooctane B ring containing the first 5 carbon atoms was synthesized in a semisynthesis starting from naturally occurring L-serine (scheme 1). This route started with conversion of the amino group of the serine methyl ester (1) to the diol ester 2 via diazotization (sodium nitrite/sulfuric acid). After protection of the primary alcohol group to a (t-buyldimethyl) TBS silyl ether (TBSCl / imidazole) and that of the secondary alcohol group with a (Bn) benzyl ether (benzyl imidate, triflic acid), the aldehyde 3 was reacted with the methyl ester of isobutyric acid (4) in an Aldol addition to alcohol 5 with 65% stereoselectivity. This group was protected as a PMB (p-methoxybenzyl) ether (again through an imidate) in 6 which enabled organic reduction of the ester to the aldehyde in 7 with DIBAL.
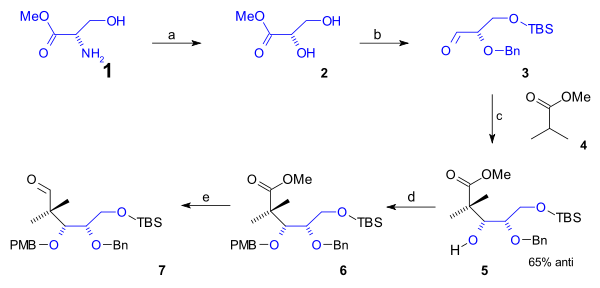
Scheme 1 Completing the cyclooctane ring required 3 more carbon atoms that were supplied by a C2 fragment in an aldol addition and a Grignard C1 fragment (scheme 2). A Mukaiyama aldol addition (magnesium bromide / toluene) took place between aldehyde 7 and ketene silyl acetal 8 with 71% stereoselectivity to alcohol 9 which was protected as the TBS ether 10 (TBSOTf, 2,6-lutidine). The ester group was reduced with DIBAL to an alcohol and then back oxidized to aldehyde 11 by Swern oxidation. Alkylation by methyl magnesium bromide to alcohol 12 and another Swern oxidation gave ketone 13. This group was converted to the silyl enol ether 14 (LHMDS, TMSCl) enabling it to react with NBS to alkyl bromide 15. The C20 methyl group was introduced as methyl iodide in a nucleophilic substitution with a strong base (LHMDS in HMPA) to bromide 16. Then in preparation to ring-closure the TBS ether was deprotected (HCl/THF) to an alcohol which was converted to the aldehyde 17 in a Swern oxidation. The ring-closing reaction was a Reformatskii reaction with Samarium(II) iodide and acetic acid to acetate 18. The stereochemistry of this particular step was of no consequence because the acetate group is dehydrated to the alkene 19 with DBU in benzene.
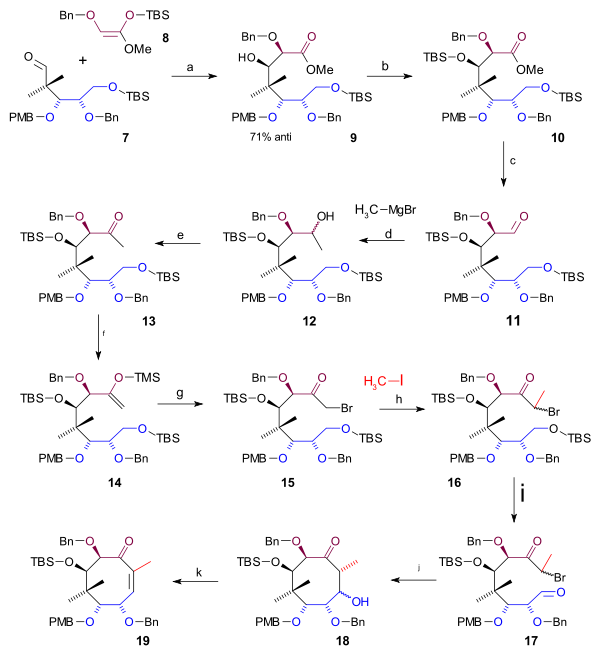
Scheme 2 Synthesis C ring
The C5 fragment 24 required for the synthesis of the C ring (scheme 3) was prepared from 2,3-dibromopropene (20) [1] by reaction with ethyl acetate (21), n-butyllithium and a copper salt, followed by organic reduction of acetate 22 to alcohol 23 (lithium aluminium hydride) and its TES silylation. Michael addition of 24 with the cyclooctane 19 to 25 with t-BuLi was catalyzed by copper cyanide. After removal of the TES group (HCl, THF), the alcohol 26 was oxidized to aldehyde 27 (TPAP, NMO)which enabled the intramolecular Aldol reaction to bicycle 28.
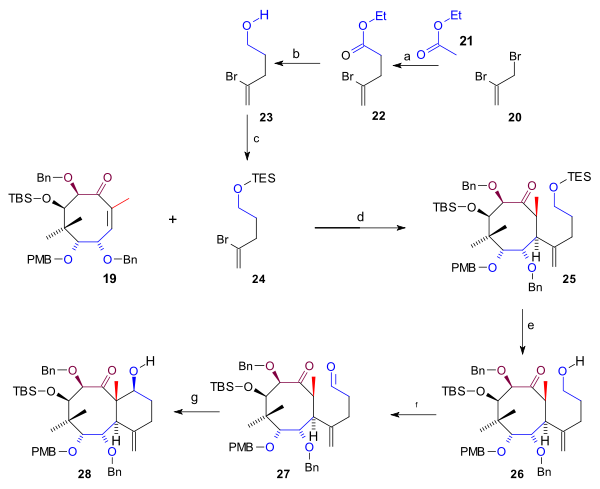
Scheme 3 Synthesis A ring
Ring A synthesis (scheme 4) started with reduction of the C9 ketone group in 28 to diol 29 with alane in toluene followed by diol protection in 30 as a dimethyl carbonate. This allowed selective oxidation of the C1 alcohol with DDQ after deprotection to ketone 31. This compound was alkylated to 32 at the C1 ketone group with the Grignard homoallyl magnesium bromide (C4 fragment completing the carbon framework) and deprotected at C11 (TBAF) to diol 33. By reaction with cyclohexylmethylsilyldichloride both alcohol groups participated in a cyclic silyl ether (34) which was again cleaved by reaction with methyl lithium exposing the C11 alcohol in 35. The A ring closure required two ketone groups for a pinacol coupling which were realized by oxidation of the C11 alcohol (TPAP, NMO) to ketone 36 and Wacker oxidation of the allyl group to diketone 37. After formation of the pinacol product 38 the benzyl groups (sodium, ammonia) and the trialkylsilyl groups (TBAF) were removed to form pentaol 39.

Scheme 4 The pentaol 39 was protected twice: two bottom hydroxyl groups as a carbonate ester (bis(trichloromethyl)carbonate, pyridine) and the C10 hydroxyl group as the acetate forming 40. The acetonide group was removed (HCl, THF), the C7 hydroxyl group protected as a TES silyl ether and the C11 OH group oxidized (TPAP, NMO) to ketone 41. The ring A diol group was next removed in a combined elimination reaction and Barton deoxygenation with 1,1'-thiocarbonyldiimidazole forming alkene 42. Finally the C15 hydroxyl group was introduced by oxidation at the allyl position with in two steps PPC and sodium acetate (to the enone) and with K-selectride to alcohol 43 which was protected as a TES ether in 44.
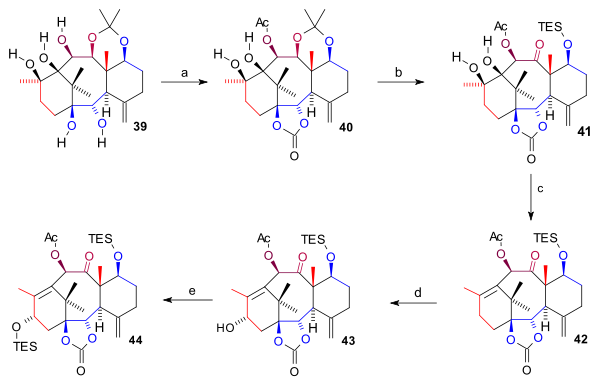
Scheme 5 Synthesis D ring
The synthesis of the D ring (scheme 6) started from 44 with allylic bromination with copper(I) bromide and benzoyl tert-butyl peroxide to bromide 45. By adding even more bromide, another bromide 46 formed (both compounds are in chemical equilibrium) with the bromine atom in an axial position. Osmium tetroxide added two hydroxyl groups to the exocyclic double bond in diol 47 and oxetane ring-closure to 48 took place with DBU in a nucleophilic substitution. Then, acylation of the C4 hydroxyl group (acetic anhydride, DMAP, pyridine) resulted in acetate 49. In the final steps phenyllithium opened the ester group to form hydroxy carbonate 50, both TES groups were removed (HF, pyr) to triol 51 (baccatin III) and the C7 hydroxyl group was back-protected to 52.
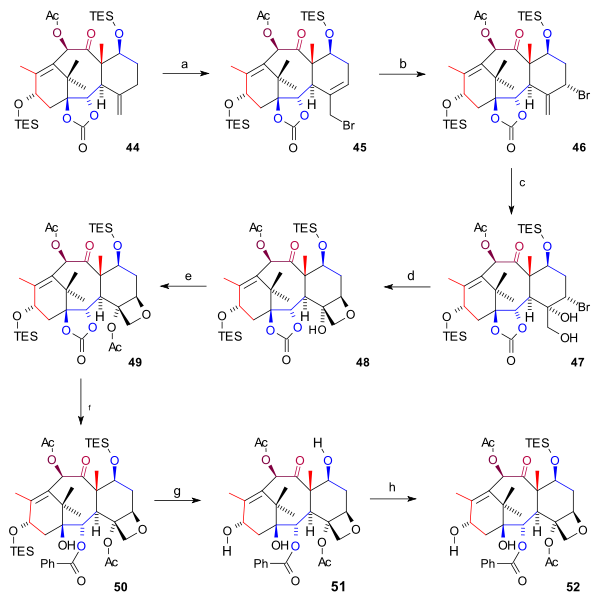
Scheme 6 Tail synthesis
The amide tail synthesis (scheme 7) was based on an asymmetric Aldol reaction. The starting compound is the commercially available Benzyloxyacetic acid 53 which was converted to the thio ester 55 (Ethanethiol) through the acid chloride 54 (thionyl chloride, pyridine). This formed the silyl enol ether 55 (n-butyllithium, trimethylsilyl chloride, Diisopropylamine) which reacted with chiral amine catalyst 58, tin triflate and nBu2(OAc)2 in a Mukaiyama aldol addition with benzaldehyde to alcohol 59 with 99% anti selectivity and 96% ee. The next step converting the alcohol group to an amine in 60 was a Mitsunobu reaction (hydrogen azide, DEAD, triphenylphosphine with azide reduction to amine by Ph3P). The amine group was benzoylated with benzoyl chloride (61) and hydrolysis removes the thioether group in 62.
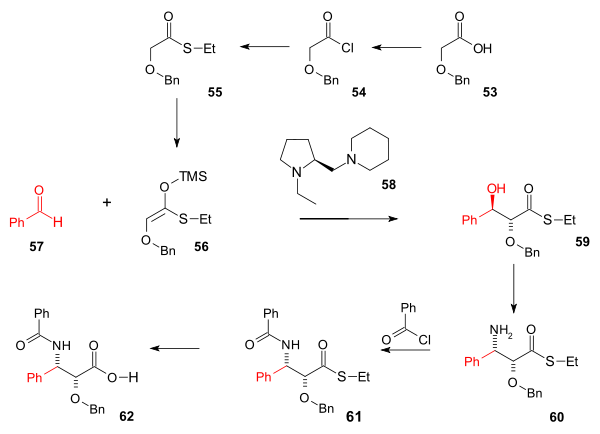
Scheme 7 Tail addition
In the final synthetic steps (scheme 8) the amide tail 62 was added to ABCD ring 52 in a esterfication catalysed by o,o'-di(2-pyridyl) thiocarbonate (DPTC) and DMAP forming ester 63. The Bn protecting group was removed by hydrogenation using palladium hydroxide on carbon (64) and finally the TES group was removed by HF and pyridine to yield Taxol 65.
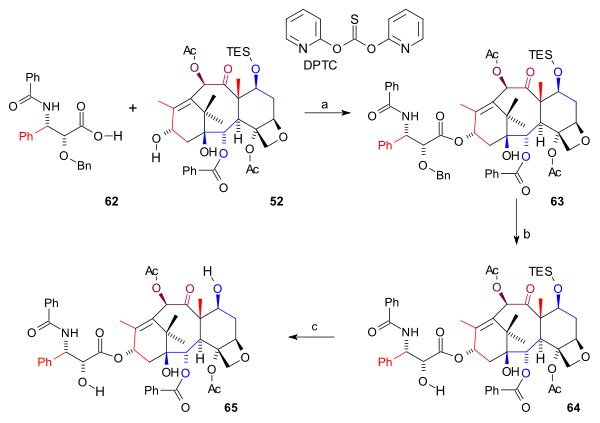
Scheme 8 see also
- Paclitaxel total synthesis
- Danishefsky Taxol total synthesis
- Holton Taxol total synthesis
- Kuwajima Taxol total synthesis
- Nicolaou Taxol total synthesis
- Wender Taxol total synthesis
External links
References
- Mukaiyama, Teruaki (1999). "Asymmetric Total Synthesis of Taxol Teruaki Mukaiyama , Isamu Shiina, Hayato Iwadare, Masahiro Saitoh, Toshihiro Nishimura, Naoto Ohkawa, Hiroki Sakoh, Koji Nishimura, Yu-ichirou Tani, Masatoshi Hasegawa, Koji Yamada , Katsuyuki Saitoh". Chem. Eur. J. 5 (1): 121–161. doi:10.1002/(SICI)1521-3765(19990104)5:1<121::AID-CHEM121>3.0.CO;2-O.
- ^ R. Lespieau and M. Bourgue (1941), "2,3-Dibromopropene", Org. Synth., http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv1p0209; Coll. Vol. 1: 209
Categories:









Wikimedia Foundation. 2010.