- Maximum parsimony (phylogenetics)
-
Parsimony is a non-parametric statistical method commonly used in computational phylogenetics for estimating phylogenies. Under parsimony, the preferred phylogenetic tree is the tree that requires the least evolutionary change to explain some observed data.
Contents
In detail
Parsimony is part of a class of character-based tree estimation methods which use a matrix of discrete phylogenetic characters to infer one or more optimal phylogenetic trees for a set of taxa, commonly a set of species or reproductively-isolated populations of a single species. These methods operate by evaluating candidate phylogenetic trees according to an explicit optimality criterion; the tree with the most favorable score is taken as the best estimate of the phylogenetic relationships of the included taxa. Maximum parsimony is used with most kinds of phylogenetic data; until recently, it was the only widely-used character-based tree estimation method used for morphological data.
Estimating phylogenies is not a trivial problem. A huge number of possible phylogenetic trees exist for any reasonably sized set of taxa; for example, a mere ten species gives over two million possible unrooted trees. These possibilities must be searched to find a tree that best fits the data according to the optimality criterion. However, the data themselves do not lead to a simple, arithmetic solution to the problem. Ideally, we would expect the distribution of whatever evolutionary characters (such as phenotypic traits or alleles) to directly follow the branching pattern of evolution. Thus we could say that if two organisms possess a shared character, they should be more closely related to each other than to a third organism that lacks this character (provided that character was not present in the last common ancestor of all three, in which case it would be a symplesiomorphy). We would predict that bats and monkeys are more closely related to each other than either is to a fish, because they both possess hair—a synapomorphy. However, we cannot say that bats and monkeys are more closely related to one another than they are to whales because they share hair, because we believe the last common ancestor of the three had hair.
However, the phenomena of convergent evolution, parallel evolution, and evolutionary reversals (collectively termed homoplasy) add an unpleasant wrinkle to the problem of estimating phylogeny. For a number of reasons, two organisms can possess a trait not present in their last common ancestor: If we naively took the presence of this trait as evidence of a relationship, we would reconstruct an incorrect tree. Real phylogenetic data include substantial homoplasy, with different parts of the data suggesting sometimes very different relationships. Methods used to estimate phylogenetic trees are explicitly intended to resolve the conflict within the data by picking the phylogenetic tree that is the best fit to all the data overall, accepting that some data simply will not fit. It is often mistakenly believed that parsimony assumes that convergence is rare; in fact, even convergently-derived characters have some value in maximum-parsimony-based phylogenetic analyses, and the prevalence of convergence does not systematically affect the outcome of parsimony-based methods.[1]
Data that do not fit a tree perfectly are not simply "noise", they can contain relevant phylogenetic signal in some parts of a tree, even if they conflict with the tree overall. In the whale example given above, the lack of hair in whales is homoplastic: It reflects a return to the condition present in ancient ancestors of mammals, who lacked hair. This similarity between whales and ancient mammal ancestors is in conflict with the tree we accept, since it implies that the mammals with hair should form a group excluding whales. However, among the whales, the reversal to hairlessness actually correctly associates the various types of whales (including dolphins and porpoises) into the group Cetacea. Still, the determination of the best-fitting tree—and thus which data do not fit the tree—is a complex process. Maximum parsimony is one method developed to do this.
Character data
The input data used in a maximum parsimony analysis is in the form of "characters" for a range of taxa. There is no generally agreed-upon definition of a phylogenetic character, but operationally a character can be thought of as an attribute, an axis along which taxa are observed to vary. These attributes can be physical (morphological), molecular, genetic, physiological, or behavioral. The only widespread agreement on characters seems to be that variation used for character analysis should reflect heritable variation. Whether it must be directly heritable, or whether indirect inheritance (e.g., learned behaviors) is acceptable, is not entirely resolved.
Each character is divided into discrete character states, into which the variations observed are classified. Character states are often formulated as descriptors, describing the condition of the character substrate. For example, the character "eye color" might have the states "blue" and "brown." Characters can have two or more states (they can have only one, but these characters lend nothing to a maximum parsimony analysis, and are often excluded).
Coding characters for phylogenetic analysis is not an exact science, and there are numerous complicating issues. Typically, taxa are scored with the same state if they are more similar to one another in that particular attribute than each is to taxa scored with a different state. This is not straightforward when character states are not clearly delineated or when they fail to capture all of the possible variation in a character. How would one score the previously mentioned character for a taxon (or individual) with hazel eyes? Or green? As noted above, character coding is generally based on similarity: Hazel and green eyes might be lumped with blue because they are more similar to that color (being light), and the character could be then recoded as "eye color: light; dark." Alternatively, there can be multi-state characters, such as "eye color: brown; hazel, blue; green."
Ambiguities in character state delineation and scoring can be a major source of confusion, dispute, and error in phylogenetic analysis using character data. Note that, in the above example, "eyes: present; absent" is also a possible character, which creates issues because "eye color" is not applicable if eyes are not present. For such situations, a "?" ("unknown") is scored, although sometimes "X" or "-" (the latter usually in sequence data) are used to distinguish cases where a character cannot be scored from a case where the state is simply unknown. Current implementations of maximum parsimony generally treat unknown values in the same manner: the reasons the data are unknown have no particular effect on analysis. Effectively, the program treats a ? as if it held the state that would involve the fewest extra steps in the tree (see below), although this is not an explicit step in the algorithm.
Genetic data are particularly amenable to character-based phylogenetic methods such as maximum parsimony because protein and nucleotide sequences are naturally discrete: A particular position in a nucleotide sequence can be either adenine, cytosine, guanine, or thymine / uracil, or a sequence gap; a position (residue) in a protein sequence will be one of the basic amino acids or a sequence gap. Thus, character scoring is rarely ambiguous, except in cases where sequencing methods fail to produce a definitive assignment for a particular sequence position. Sequence gaps are sometimes treated as characters, although there is no consensus on how they should be coded.
Characters can be treated as unordered or ordered. For a binary (two-state) character, this makes little difference. For a multi-state character, unordered characters can be thought of as having an equal "cost" (in terms of number of "evolutionary events") to change from any one state to any other; complementarily, they do not require passing through intermediate states. Ordered characters have a particular sequence in which the states must occur through evolution, such that going between some states requires passing through an intermediate. This can be thought of complementarily as having different costs to pass between different pairs of states. In the eye-color example above, it is possible to leave it unordered, which imposes the same evolutionary "cost" to go from brown-blue, green-blue, green-hazel, etc. Alternatively, it could be ordered brown-hazel-green-blue; this would normally imply that it would cost two evolutionary events to go from brown-green, three from brown-blue, but only one from brown-hazel. This can also be thought of as requiring eyes to evolve through a "hazel stage" to get from brown to green, and a "green stage" to get from hazel to blue, etc.
There is a lively debate on the utility and appropriateness of character ordering, but no consensus. Some authorities order characters when there is a clear logical, ontogenetic, or evolutionary transition among the states (for example, "legs: short; medium; long"). Some accept only some of these criteria. Some run an unordered analysis, and order characters that show a clear order of transition in the resulting tree (which practice might be accused of circular reasoning). Some authorities refuse to order characters at all, suggesting that it biases an analysis to require evolutionary transitions to follow a particular path.
It is also possible to apply differential weighting to individual characters. This is usually done relative to a "cost" of 1. Thus, some characters might be seen as more likely to reflect the true evolutionary relationships among taxa, and thus they might be weighted at a value 2 or more; changes in these characters would then count as two evolutionary "steps" rather than one when calculating tree scores (see below). There has been much discussion in the past about character weighting. Most authorities now weight all characters equally, although exceptions are common. For example, allele frequency data is sometimes pooled in bins and scored as an ordered character. In these cases, the character itself is often downweighted so that small changes in allele frequencies count less than major changes in other characters. Also, the third codon position in a coding nucleotide sequence is particularly labile, and is sometimes downweighted, or given a weight of 0, on the assumption that it is more likely to exhibit homoplasy. In some cases, repeated analyses are run, with characters reweighted in inverse proportion to the degree of homoplasy discovered in the previous analysis (termed successive weighting); this is another technique that might be considered circular reasoning.
Character state changes can also be weighted individually. This is often done for nucleotide sequence data; it has been empirically determined that certain base changes (A-C, A-T, G-C, G-T, and the reverse changes) occur much less often than others. These changes are therefore often weighted more. As shown above in the discussion of character ordering, ordered characters can be thought of as a form of character state weighting.
Some systematists prefer to exclude characters known to be, or suspected to be, highly homoplastic or that have a large number of unknown entries ("?"). As noted below, theoretical and simulation work has demonstrated that this is likely to sacrifice accuracy rather than improve it. This is also the case with characters that are variable in the terminal taxa: theoretical, congruence, and simulation studies have all demonstrated that such polymorphic characters contain significant phylogenetic information.[citation needed]
Taxon sampling
The time required for a parsimony analysis (or any phylogenetic analysis) is proportional to the number of taxa (and characters) included in the analysis. Also, because more taxa require more branches to be estimated, more uncertainty may be expected in large analyses. Because data collection costs in time and money often scale directly with the number of taxa included, most analyses include only a fraction of the taxa that could have been sampled. Indeed, some authors have contended that four taxa (the minimum required to produce a meaningful unrooted tree) are all that is necessary for accurate phylogenetic analysis, and that more characters are more valuable than more taxa in phylogenetics. This has led to a raging controversy about taxon sampling.
Empirical, theoretical, and simulation studies have led to a number of dramatic demonstrations of the importance of adequate taxon sampling. Most of these can be summarized by a simple observation: a phylogenetic data matrix has dimensions of characters times taxa. Doubling the number of taxa doubles the amount of information in a matrix just as surely as doubling the number of characters. Each taxon represents a new sample for every character, but, more importantly, it (usually) represents a new combination of character states. These character states can not only determine where that taxon is placed on the tree, they can inform the entire analysis, possibly causing different relationships among the remaining taxa to be favored by changing estimates of the pattern of character changes.
The most disturbing weakness of parsimony analysis, that of long-branch attraction (see below) is particularly pronounced with poor taxon sampling, especially in the four-taxon case. This is a well-understood case in which additional character sampling may not improve the quality of the estimate. As taxa are added, they often break up long branches (especially in the case of fossils), effectively improving the estimation of character state changes along them. Because of the richness of information added by taxon sampling, it is even possible to produce highly accurate estimates of phylogenies with hundreds of taxa using only a few thousand characters.[citation needed]
Although many studies have been performed, there is still much work to be done on taxon sampling strategies. Because of advances in computer performance, and the reduced cost and increased automation of molecular sequencing, sample sizes overall are on the rise, and studies addressing the relationships of hundreds of taxa (or other terminal entities, such as genes) are becoming common. Of course, this is not to say that adding characters is not also useful; the number of characters is increasing as well.
Some systematists prefer to exclude taxa based on the number of unknown character entries ("?") they exhibit, or because they tend to "jump around" the tree in analyses (i.e., they are "wildcards"). As noted below, theoretical and simulation work has demonstrated that this is likely to sacrifice accuracy rather than improve it. Although these taxa may generate more most-parsimonious trees (see below), methods such as agreement subtrees and reduced consensus can still extract information on the relationships of interest.
It has been observed that inclusion of more taxa tends to lower overall support values (bootstrap percentages or decay indices, see below). The cause of this is clear: as additional taxa are added to a tree, they subdivide the branches to which they attach, and thus dilute the information that supports that branch. While support for individual branches is reduced, support for the overall relationships is actually increased. Consider analysis that produces the following tree: (fish ,(lizard ,(whale, (cat, monkey)))). Adding a rat and a walrus will probably reduce the support for the (whale, (cat, monkey)) clade, because the rat and the walrus may fall within this clade, or outside of the clade, and since these five animals are all relatively closely related, there should be more uncertainty about their relationships. Within error, it may be impossible to determine any of these animals' relationships relative to one another. However, the rat and the walrus will probably add character data that cements the grouping any two of these mammals exclusive of the fish or the lizard; where the initial analysis might have been misled, say, by the presence of fins in the fish and the whale, the presence of the walrus, with blubber and fins like a whale but whiskers like a cat and a rat, firmly ties the whale to the mammals.
To cope with this problem, agreement subtrees, reduced consensus, and double-decay analysis seek to identify supported relationships (in the form of "n-taxon statements," such as the four-taxon statement "(fish, (lizard, (cat, whale)))") rather than whole trees. If the goal of an analysis is a resolved tree, as is the case for comparative phylogenetics, these methods cannot solve the problem. However, if the tree estimate is so poorly supported, the results of any analysis derived from the tree will probably be too suspect to use anyway.
Analysis
A maximum parsimony analysis runs in a very straightforward fashion. Trees are scored according to the degree to which they imply a parsimonious distribution of the character data. The most parsimonious tree for the dataset represents the preferred hypothesis of relationships among the taxa in the analysis.
Trees are scored (evaluated) by using a simple algorithm to determine how many "steps" (evolutionary transitions) are required to explain the distribution of each character. A step is, in essence, a change from one character state to another, although with ordered characters some transitions require more than one step. Contrary to popular belief, the algorithm does not explicitly assign particular character states to nodes (branch junctions) on a tree: the least number of steps can involve multiple, equally costly assignments and distributions of evolutionary transitions. What is optimized is the total number of changes.
There are many more possible phylogenetic trees than can be searched exhaustively for more than eight taxa or so. A number of algorithms are therefore used to searching among the possible trees. Many of these involve taking an initial tree (usually the favored tree from the last iteration of the algorithm), and perturbing it to see if the change produces a higher score.
The trees resulting from parsimony search are unrooted: They show all the possible relationships of the included taxa, but they lack any statement on relative times of divergence. A particular branch is chosen to root the tree by the user. This branch is then taken to be outside all the other branches of the tree, which together form a monophyletic group. This imparts a sense of relative time to the tree. Incorrect choice of a root can result in incorrect relationships on the tree, even if the tree is itself correct in its unrooted form.
Parsimony analysis often returns a number of equally most-parsimonious trees (MPTs). A large number of MPTs is often seen as an analytical failure, and is widely believed to be related to the number of missing entries ("?") in the dataset, characters showing too much homoplasy, or the presence of topologically labile "wildcard" taxa (which may have many missing entries). Numerous methods have been proposed to reduce the number of MPTs, including removing characters or taxa with large amounts of missing data before analysis, removing or downweighting highly homoplastic characters (successive weighting) or removing wildcard taxa (the phylogenetic trunk method) a posteriori and then reanalyzing the data.
Numerous theoretical and simulation studies have demonstrated that highly homoplastic characters, characters and taxa with abundant missing data, and "wildcard" taxa contribute to the analysis. Although excluding characters or taxa may appear to improve resolution, the resulting tree is based on less data, and is therefore a less reliable estimate of the phylogeny (unless the characters or taxa are non informative, see safe taxonomic reduction). Today's general consensus is that having multiple MPTs is a valid analytical result; it simply indicates that there is insufficient data to resolve the tree completely. In many cases, there is substantial common structure in the MPTs, and differences are slight and involve uncertainty in the placement of a few taxa. There are a number of methods for summarizing the relationships within this set, including consensus trees, which show common relationships among all the taxa, and pruned agreement subtrees, which show common structure by temporarily pruning "wildcard" taxa from every tree until they all agree. Reduced consensus takes this one step further, but showing all subtrees (and therefore all relationships) supported by the input trees.
Even if multiple MPTs are returned, parsimony analysis still basically produces a point-estimate, lacking confidence intervals of any sort. This has often been levelled as a criticism, since there is certainly error in estimating the most-parsimonious tree, and the method does not inherently include any means of establishing how sensitive its conclusions are to this error. Several methods have been used to assess support.
Jackknifing and bootstrapping, well-known statistical resampling procedures, have been employed with parsimony analysis. The jackknife, which involves resampling without replacement ("leave-one-out") can be employed on characters or taxa; interpretation may become complicated in the latter case, because the variable of interest is the tree, and comparison of trees with different taxa is not straightforward. The bootstrap, resampling with replacement (sample x items randomly out of a sample of size x, but items can be picked multiple times), is only used on characters, because adding duplicate taxa does not change the result of a parsimony analysis. The bootstrap is much more commonly employed in phylogenetics (as elsewhere); both methods involve an arbitrary but large number of repeated iterations involving perturbation of the original data followed by analysis. The resulting MPTs from each analysis are pooled, and the results are usually presented on a 50% Majority Rule Consensus tree, with individual branches (or nodes) labelled with the percentage of bootstrap MPTs in which they appear. This "bootstrap percentage" (which is not a P-value, as is sometimes claimed) is used as a measure of support. Technically, it is supposed to be a measure of repeatability, the probability that that branch (node, clade) would be recovered if the taxa were sampled again. Experimental tests with viral phylogenies suggest that the bootstrap percentage is not a good estimator of repeatability for phylogenetics, but it is a reasonable estimator of accuracy.[citation needed] In fact, it has been shown that the bootstrap percentage, as an estimator of accuracy, is biased, and that this bias results on average in an underestimate of confidence (such that as little as 70% support might really indicate up to 95% confidence). However, the direction of bias cannot be ascertained in individual cases, so assuming that high values bootstrap support indicate even higher confidence is unwarranted.
Another means of assessing support is Bremer support, or the decay index (which is technically not an index). This is simply the difference in number of steps between the score of the MPT(s), and the score of the most parsimonious tree that does NOT contain a particular clade (node, branch). It can be thought of as the number of steps you have to add to lose that clade; implicitly, it is meant to suggest how great the error in the estimate of the score of the MPT must be for the clade to no longer be supported by the analysis, although this is not necessarily what it does. Decay index values are often fairly low (one or two steps being typical), but they often appear to be proportional to bootstrap percentages. However, interpretation of decay values is not straightforward, and they seem to be preferred by authors with philosophical objections to the bootstrap (although many morphological systematists, especially paleontologists, report both). Double-decay analysis is a decay counterpart to reduced consensus that evaluates the decay index for all possible subtree relationships (n-taxon statements) within a tree.
Problems with maximum parsimony phylogeny estimation
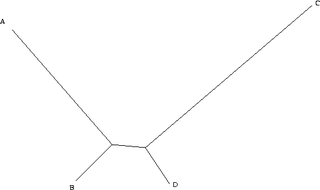 An example of long branch attraction. Branches A & C have a high number of substitutions.
An example of long branch attraction. Branches A & C have a high number of substitutions.
Maximum parsimony is a very simple approach, and is popular for this reason. However, it is not statistically consistent. That is, it is not guaranteed to produce the true tree with high probability, given sufficient data. Consistency, here meaning the monotonic convergence on the correct answer with the addition of more data, is a desirable property of any statistical method. As demonstrated in 1978 by Joe Felsenstein,[2] maximum parsimony can be inconsistent under certain conditions. The category of situations in which this is known to occur is called long branch attraction, and occurs, for example, where there are long branches (a high level of substitutions) for two characters (A & C), but short branches for another two (B & D). A and B diverged from a common ancestor, as did C and D.
Assume for simplicity that we are considering a single binary character (it can either be + or -). Because the distance from B to D is small, in the vast majority of all cases, B and D will be the same. Here, we will assume that they are both + (+ and - are assigned arbitrarily and swapping them is only a matter of definition). If this is the case, there are four remaining possibilities. A and C can both be +, in which case all taxa are the same and all the trees have the same length. A can be + and C can be -, in which case only one character is different, and we cannot learn anything, as all trees have the same length. Similarly, A can be - and C can be +. The only remaining possibility is that A and C are both -. In this case, however, we group A and C together, and B and D together. As a consequence, when we have a tree of this type, the more data we collect (i.e. the more characters we study), the more we tend towards the wrong tree.
A simple and effective method for determining whether or not long branch attraction is affecting tree topology is the SAW method, named for Siddal and Whiting. If long branch attraction is suspected in a pair of taxa (A and B), simply remove taxon A ("saw" off the branch) and re-run the analysis. Then remove A and replace B, running the analysis again. If either of the taxa appear at different branch points in the absence of the other, there is evidence of long branch attraction. Since long branches can't possibly attract one another when only one is in the analysis, consistent taxon placement between treatments would indicate long branch attraction is not a problem.[3]
Several other methods of phylogeny estimation are available, including maximum likelihood, Bayesian phylogeny inference, neighbour joining, and quartet methods. Of these, the first two both use a likelihood function, and, if used properly, are theoretically immune to long-branch attraction. These methods are both parametric, meaning that they rely on an explicit model of character evolution. It has been shown that, for some suboptimal models, these methods can also be inconsistent.[citation needed]
Another complication with maximum parsimony is that finding the most parsimonious tree is an NP-Hard problem.[4] The only currently available, efficient way of obtaining a solution, given an arbitrarily large set of taxa, is by using heuristic methods which do not guarantee that the most parsimonious tree will be recovered. These methods employ hill-climbing algorithms to progressively approach the best tree. However, it has been shown that there can be "tree islands" of suboptimal solutions, and the analysis can become trapped in these local optima. Thus, complex, flexible heuristics are required to ensure that tree space has been adequately explored. Several heuristics are available, including nearest neighbor interchange (NNI), tree bisection / reconnection (TBR), and the phylogenetic ratchet. This problem is certainly not unique to MP; any method that uses an optimality criterion faces the same problem, and none offer easy solutions.
Criticism
It has been asserted that a major problem, especially for paleontology, is that maximum parsimony assumes that the only way two species can share the same nucleotide at the same position is if they are genetically related. This asserts that phylogenetic applications of parsimony assume that all similarity is homologous (other interpretations, such as the assertion that two organisms might NOT be related at all, are nonsensical). This is emphatically not the case: as with any form of character-based phylogeny estimation, parsimony is used to test the homologous nature of similarities by finding the phylogenetic tree which best accounts for all of the similarities.
For example, birds and bats have wings, while crocodiles and humans do not. If these were the only data available, maximum parsimony would tend to group crocodiles with humans, and birds with bats (as would any other method of phylogenetic inference). We believe that humans are actually more closely related to bats than to crocodiles or birds. Our belief is founded on additional data that were not considered in the one-character example (using wings). If even a tiny fraction of these additional data, including information on skeletal structure, soft-tissue morphology, integument, behaviour, genetics, etc., were included in the analysis, the faint phylogenetic signal produced by the presence of wings in birds and bats would be overwhelmed by the preponderance of data supporting the (human, bat)(bird, crocodile) tree.
It is often stated that parsimony is not relevant to phylogenetic inference because "evolution is not parsimonious." In most cases, there is no explicit alternative proposed; if no alternative is available, any statistical method is preferable to none at all. Additionally, it is not clear what would be meant if the statement "evolution is parsimonious" were in fact true. This could be taken to mean that more character changes may have occurred historically than are predicted using the parsimony criterion. Because parsimony phylogeny estimation reconstructs the minimum number of changes necessary to explain a tree, this is quite possible. However, it has been shown through simulation studies, testing with known in vitro viral phylogenies, and congruence with other methods, that the accuracy of parsimony is in most cases not compromised by this. Parsimony analysis uses the number of character changes on trees to choose the best tree, but it does not require that exactly that many changes, and no more, produced the tree. As long as the changes that have not been accounted for are randomly distributed over the tree (a reasonable null expectation), the result should not be biased. In practice, the technique is robust: maximum parsimony exhibits minimal bias as a result of choosing the tree with the fewest changes.
An analogy can be drawn with choosing among contractors based on their initial (nonbinding) estimate of the cost of a job. The actual finished cost is very likely to be higher than the estimate. Despite this, choosing the contractor who furnished the lowest estimate should theoretically result in the lowest final project cost. This is because, in the absence of other data, we would assume that all of the relevant contractors have the same risk of cost overruns. In practice, of course, unscrupulous business practices may bias this result; in phylogenetics, too, some particular phylogenetic problems (for example, long branch attraction, described above) may potentially bias results. In both cases, however, there is no way to tell if the result is going to be biased, or the degree to which it will be biased, based on the estimate itself. With parsimony too, there is no way to tell that the data are positively misleading, without comparison to other evidence.
Along the same lines, parsimony is often characterized as implicitly adopting the philosophical position that evolutionary change is rare, or that homoplasy (convergence and reversal) is minimal in evolution. This is not entirely true: parsimony minimizes the number of convergences and reversals that are assumed by the preferred tree, but this may result in a relatively large number of such homoplastic events. It would be more appropriate to say that parsimony assumes only the minimum amount of change implied by the data. As above, this does not require that these were the only changes that occurred; it simply does not infer changes for which there is no evidence. The shorthand for describing this is that "parsimony minimizes assumed homoplasies, it does not assume that homoplasy is minimal."
Parsimony is also sometimes associated with the notion that "the simplest possible explanation is the best," a generalisation of Occam's Razor. Parsimony does prefer the solution that requires the least number of unsubstantiated assumptions and unsupportable conclusions, the solution that goes the least theoretical distance beyond the data. This is a very common approach to science, especially when dealing with systems that are so complex as to defy simple models. Parsimony does not by any means necessarily produce a "simple" assumption. Indeed, as a general rule, most character datasets are so "noisy" that no truly "simple" solution is possible.
Alternatives
There are several other methods for inferring phylogenies based on discrete character data. Each offers potential advantages and disadvantages. Most of these methods have particularly avid proponents and detractors; parsimony especially has been advocated as philosophically superior (most notably by ardent cladists).
Maximum likelihood
Main article: Maximum likelihoodAmong the most popular alternative phylogenetic methods is maximum likelihood phylogenetic inference, sometimes simply called "likelihood" or "ML." Maximum likelihood is an optimality criterion, as is parsimony. Mechanically, maximum likelihood analysis functions much like parsimony analysis, in that trees are scored based on a character dataset, and the tree with the best score is selected. Maximum likelihood is a parametric statistical method, in that it employs an explicit model of character evolution. Such methods are potentially much more powerful than non-parametric statistical methods like parsimony, but only if the model used is a reasonable approximation of the processes that produced the data. Maximum likelihood has probably surpassed parsimony in popularity with nucleotide sequence data, and Bayesian phylogenetic inference, which uses the likelihood function, is becoming almost as prevalent.
Likelihood is the relative counterpart to absolute probability. If we know the number of possible outcomes of a test (N), and we know the number of those outcomes that fit a particular criterion (n), we can say that the probability of that criterion being met by an execution of that test is n/N. Thus, the probability of heads in the toss of a fair coin is 50% (1/2). What if we don't know the number of possible outcomes? Obviously, we cannot then calculate probabilities. However, if we observe that one outcome happens twice as often as the other over an arbitrarily large number of tests, we can say that that outcome is twice as likely. Likelihoods are proportional to the true probabilities: if an outcome is twice as likely, we can say that it is twice as probable, even though we cannot say how probable it is.
Practically, the probability of a tree cannot be calculated directly. The probability of the data given a tree can be calculated if you assume a specific set of probabilities of character change (a model). The critical part of likelihood analysis is that the probability of the data given the tree is the likelihood of the tree given the data. Thus, the tree that has the highest probability of producing the observed data is the most likely tree.
Maximum likelihood, as implemented in phylogenetics, uses a stochastic model that gives the probability of a particular character changing at any given point on a tree. This model can have a potentially large number of parameters, which can account for differences in the probabilities of particular states, the probabilities of particular changes, and differences in the probabilities of change among characters.
A likelihood tree has meaningful branch lengths (i.e. it is a phylogram); these lengths are usually interpreted as being proportional to the average probability of change for characters on that branch (thus, on a branch of length 1, we would expect an average of one change per character, which is a lot). The state of each character is plotted on the tree, and the probability of that distribution of character states is calculated using the model and the branch lengths (which can be altered to maximize the probability of the data). This is the probability of that character, given the tree. The probabilities of all of the characters is multiplied together; they are usually negative log-transformed and added (producing the same effect), because the numbers become very small very quickly. This sum is the probability of the data, given the tree, or the likelihood of the tree. The tree with the highest likelihood (lowest negative log-transformed likelihood) given the data is preferred.
In the above analogy regarding choosing a contractor, maximum likelihood would be analogous to gathering data on the final cost of broadly comparable jobs performed by each contractor over the past year, and selecting the contractor with the lowest average cost for those comparable jobs. This method would be highly dependent on how comparable the jobs are, but, if they are properly chosen, it will produce a better estimate of the actual cost of the job. Further, it would not be misled by bias in contractor estimates, because it is based on the final cost, not on the (potentially biased) estimates.
In practice, maximum likelihood tends to favor trees that are very similar to the most parsimonious tree(s) for the same dataset. It has been shown to outperform parsimony in certain situations where the latter is known to be biased, including long-branch attraction. Note, however, that the performance of likelihood is dependent on the quality of the model employed; an incorrect model can produce a biased result. Studies have shown that incorporating a parameter to account for differences in rate of evolution among characters is often critical to accurate estimation of phylogenies; failure to model this or other crucial parameters may produce incorrect or biased results. Model parameters are usually estimated from the data, and the number (and type) of parameters is often determined using the hierarchical likelihood ratio test. The consequences of mis-specified models are just beginning to be explored in detail.
Likelihood is generally regarded as a more desirable method than parsimony, in that it is statistically consistent, and has a better statistical foundation, and because it allows complex modeling of evolutionary processes. A major drawback is that ML is still quite slow relative to parsimony methods, sometimes requiring days to run large datasets. Maximum likelihood phylogenetic inference was proposed in the mid-Twentieth Century, but it has only been a popular method for phylogenetic inference since the 1990s, when computational power caught up with tremendous demands of ML analysis. Newer algorithms and implementations are bringing analysis times for large datasets into acceptable ranges. Until these methods gain widespread acceptance, parsimony will probably be preferred for extremely large datasets, especially when bootstrapping is used to assess confidence in the results.
One area where parsimony still holds much sway is in the analysis of morphological data. Until recently, stochastic models of character change were not available for non-molecular data. New methods, proposed by Paul Lewis, make essentially the same assumptions that parsimony analysis does, but do so within a likelihood framework. These models are not, however, widely implemented, and, unless modified, they require the modification of existing datasets (to deal with ordered characters, and the tendency to not record autapomorphies in morphological datasets.
Maximum likelihood has been criticised as assuming neutral evolution implicitly in its adoption of a stochastic model of evolution. This is not necessarily the case: as with parsimony, assuming a stochastic model does not presume that all evolution is stochastic. In practice, likelihood is robust to deviations from stochasticity. It performs well even on coding sequences that include sites believed to be under selection.
A related objection (often brought up by parsimony-only advocates) is the idea that evolution is too complex or too poorly understood to be modeled. This objection probably rests on a misunderstanding of the term "model." While it is customary to think of models as representing the mechanics of a process, this is not necessarily literally the case. In fact, a model is often selected not so much for its faithful reproduction of the phenomenon as its ability to make predictions. In practice, it is best not to try and exactly fit a model to a process, because there is a trade-off between number of parameters in a model and its statistical power. Stochasticity may be a reasonably good fit to evolutionary data at a broad level, even if it does not accurately mirror the process at finer scales.
By analogy, no one claims that the human foot varies only in length and width, but differing combinations of length and width values can be combined to fit a wide variety of feet. In some cases, a slightly wider overall foot may be better fitted by increasing overall size rather than instep width, while a foot with a narrower heel might be better fit by a wider instep and a smaller shoe. Adding several more measurements would probably improve shoe fit somewhat, but would be impractical from a business standpoint. With increasingly precise fitting, differences between feet would make selling matched pairs of shoes impossible, and differences through time would mean that a proper fit at purchase might not be a proper fit when worn.
Parsimony has recently been shown to be more likely to recover the true tree in the face of profound changes in evolutionary ("model") parameters (e.g., the rate of evolutionary change) within a tree (Kolaczkowski & Thornton 2004). This is particularly troublesome, since it is generally agreed that such changes may be a significant feature of deep divergences. Likelihood has had substantial success recovering known in vitro viral phylogenies, simulated phylogenies, and phylogenies confirmed by other method. It seems likely therefore that this potential complication does not strongly bias results for more shallow divergences. Several research groups are currently exploring ways to incorporate profound shifts in evolutionary parameters into likelihood analysis.
Bayesian phylogenetic inference
Main article: Bayesian inference in phylogenyBayesian phylogenetics uses the likelihood function, and is normally implemented using the same models of evolutionary change used in Maximum Likelihood. It is very different, however, in both theory and application. Bayesian phylogenetic analysis uses Bayes' theorem, which relates the posterior probability of a tree to the likelihood of data, and the prior probability of the tree and model of evolution. However, unlike parsimony and likelihood methods, Bayesian analysis does not produce a single tree or set of equally optimal trees. Bayesian analysis uses the likelihood of trees in a Markov chain Monte Carlo (MCMC) simulation to sample trees in proportion to their likelihood, thereby producing a credible sample of trees.
One commonly cited drawback of Bayesian analysis is the need to explicitly set out a set of prior probabilities for the range of potential outcomes. The idea of incorporating prior probabilities into an analysis has been suggested as a potential source of bias. Bayesian methods involve other potential issues, such as the evaluation of "convergence," the point at which the MCMC process stops searching for the "space" of credible solutions and begins to build the credible sample.
Distance matrix methods
Main article: Distance matrices in phylogenyNon-parametric distance methods were originally applied to phenetic data using a matrix of pairwise distances. These distances are then reconciled to produce a tree (a phylogram, with informative branch lengths). The distance matrix can come from a number of different sources, including measured distance (for example from immunological studies) or morphometric analysis, various pairwise distance formulae (such as euclidean distance) applied to discrete morphological characters, or genetic distance from sequence, restriction fragment, or allozyme data. For phylogenetic character data, raw distance values can be calculated by simply counting the number of pairwise differences in character states (Manhattan distance).
Several simple algorithms exist to construct a tree directly from pairwise distances, including UPGMA and neighbor joining (NJ), but these will not necessarily produce the best tree for the data. UPGMA assumes an ultrametric tree (a tree where all the path-lengths from the root to the tips are equal). Neighbor-joining is a form of star decomposition, and can very quickly produce reasonable trees. It is very often used on its own, and in fact quite frequently produces reasonable trees.
Phylogeny estimation using distance methods has produced a number of controversies. The relationship between individual characters and the tree is lost in the process of reducing characters to distances. Since these methods do not use character data directly, and information locked in the distribution of character states can be lost in the pairwise comparisons. Also, some complex phylogenetic relationships may produce biased distances. Despite these potential problems, distance methods are extremely fast, and they often produce a reasonable estimate of phylogeny. They also have certain benefits over the methods that use characters directly. Notably, distance methods allow use of data that may not be easily converted to character data, such as DNA-DNA hybridization assays.
References
- ^ Sober, E. (1983). "Parsimony in Systematics: Philosophical Issues". Annual Review of Ecology and Systematics 14: 335–357. doi:10.1146/annurev.es.14.110183.002003.
- ^ J. Felsenstein (1978). "Cases in which parsimony and compatibility methods will be positively misleading.". Systematic Zoology 27: 401–410. doi:10.1093/sysbio/27.4.401.
- ^ Siddall, M. E.; Whiting, M. F. (1999). "Long-Branch Abstractions". Cladistics 15: 9–24. doi:10.1111/j.1096-0031.1999.tb00391.x.
- ^ William H. E. Day, Computational complexity of inferring phylogenies from dissimilarity matrices. Bulletin of Mathematical Biology, volume 49, number 4, pages 461-467.
- Bremer, K.�r. (1994). "Branch Support and Tree Stability". Cladistics 10 (3): 295–304. doi:10.1111/j.1096-0031.1994.tb00179.x.
- Kolaczkowski, B; Thornton, JW (2004). "Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous". Nature 431 (7011): 980–984. doi:10.1038/nature02917. PMID 15496922..
Topics in phylogenetics Relevant fields Basic concepts Inference methods Maximum parsimony · Maximum likelihood · Neighbor-joining · UPGMA · Bayesian inference · Least squares · Three-taxon analysisCurrent topics -morphy -phyly List of evolutionary biology topicsCategories:
Wikimedia Foundation. 2010.