- Orbital hybridisation
-
 Four sp3 orbitals.
Four sp3 orbitals.
 Three sp2 orbitals.
Three sp2 orbitals.In chemistry, hybridisation (or hybridization) is the concept of mixing atomic orbitals to form new hybrid orbitals suitable for the qualitative description of atomic bonding properties. Hybridised orbitals are very useful in the explanation of the shape of molecular orbitals for molecules. It is an integral part of valence bond theory. Although sometimes taught together with the valence shell electron-pair repulsion (VSEPR) theory, valence bond and hybridization are in fact not related to the VSEPR model.[1]
Contents
Historical development
Chemist Linus Pauling first developed the hybridisation theory in order to explain the structure of molecules such as methane (CH4).[2] This concept was developed for such simple chemical systems, but the approach was later applied more widely, and today it is considered an effective heuristic for rationalizing the structures of organic compounds.
Hybridisation theory is not as practical for quantitative calculations as molecular orbital theory. Problems with hybridisation are especially notable when the d orbitals are involved in bonding, as in coordination chemistry and organometallic chemistry. Although hybridisation schemes in transition metal chemistry can be used, they are not generally as accurate.
Orbitals are a model representation of the behaviour of electrons within molecules. In the case of simple hybridisation, this approximation is based on atomic orbitals, similar to those obtained for the hydrogen atom, the only atom for which an exact analytic solution to its Schrödinger equation is known. In heavier atoms, like carbon, nitrogen, and oxygen, the atomic orbitals used are the 2s and 2p orbitals, similar to excited state orbitals for hydrogen. Hybridised orbitals are assumed to be mixtures of these atomic orbitals, superimposed on each other in various proportions. The theory of hybridisation is most applicable under these assumptions. It gives a simple orbital picture equivalent to Lewis structures. Hybridisation is not required to describe molecules, but for molecules made up from carbon, nitrogen and oxygen (and to a lesser extent, sulfur and phosphorus) the hybridisation theory/model makes the description much easier.
The hybridisation theory finds its use mainly in organic chemistry. Its explanation starts with the way bonding is organized in methane.
Types of Hybridisations
sp3 hybrids
Hybridisation describes the bonding atoms from an atom's point of view. That is, for a tetrahedrally coordinated carbon (e.g., methane, CH4), the carbon should have 4 orbitals with the correct symmetry to bond to the 4 hydrogen atoms. The problem with the existence of methane is now this: carbon's ground state configuration is 1s2 2s2 2px1 2py1 or more easily read:

The valence bond theory would predict, based on the existence of two half-filled p-type orbitals (the designations px py or pz are meaningless at this point, as they do not fill in any particular order), that C forms two covalent bonds, i.e., CH2 (methylene). However, methylene is a very reactive molecule (see also: carbene) and cannot exist outside of a molecular system. Therefore, this theory alone cannot explain the existence of CH4.
Furthermore, ground state orbitals cannot be used for bonding in CH4. While exciting a 2s electron into a 2p orbital would, in theory, allow for four bonds according to the valence bond theory, (which has been proved experimentally correct for systems like O2) this would imply that the various bonds of CH4 would have differing energies due to differing levels of orbital overlap. Once again, this has been experimentally disproved: any hydrogen can be removed from a carbon with equal ease.
To summarise, to explain the existence of CH4 (and many other molecules) a method by which as many as 12 bonds (for transition metals) of equal strength (and therefore equal length) was required.
The first step in hybridisation is the excitation of one (or more) electrons (we consider the carbon atom in methane, for simplicity of the discussion):

The proton that forms the nucleus of a hydrogen atom attracts one of the lower-energy valence electrons on carbon. This causes an excitation, moving a 2s electron into a 2p orbital. This, however, increases the influence of the carbon nucleus on the valence electrons by increasing the effective core potential (the amount of charge the nucleus exerts on a given electron = Charge of Core − Charge of all electrons closer to the nucleus). The effective core potential is also known as the effective nuclear charge, or Zeff.
The solution to the Schrödinger equation for this configuration is a linear combination of the s and p wave functions, or orbitals, known as a hybridized orbital.[3] In the case of carbon attempting to bond with four hydrogens, four orbitals are required. Therefore, the 2s orbital (core orbitals are almost never involved in bonding) "mixes" with the three 2p orbitals to form four sp3 hybrids (read as s-p-three). See graphical summary below.
becomes

In CH4, four sp3 hybridised orbitals are overlapped by hydrogen's 1s orbital, yielding four σ (sigma) bonds (that is, four single covalent bonds). The four bonds are of the same length and strength. This theory fits our requirements.
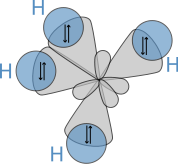 translates into
translates into 
An alternative view is: View the carbon as the C4− anion. In this case all the orbitals on the carbon are filled:

If we now recombine these orbitals with the empty s-orbitals of 4 hydrogens (4 protons, H+) and then allow maximum separation between the 4 hydrogens (i.e., tetrahedral surrounding of the carbon), we see that at any orientation of the p-orbitals, a single hydrogen has an overlap of 25% with the s-orbital of the C, and a total of 75% of overlap with the 3 p-orbitals (see that the relative percentages are the same as the character of the respective orbital in an sp3-hybridisation model, 25% s- and 75% p-character).
According to the orbital hybridisation theory, the valence electrons in methane should be equal in energy but its photoelectron spectrum[4] shows two bands, one at 12.7 eV (one electron pair) and one at 23 eV (three electron pairs). This apparent inconsistency can be explained when one considers additional orbital mixing taking place when the sp3 orbitals mix with the 4 hydrogen orbitals.
sp2 hybrids
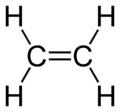 Ethene structure
Ethene structureOther carbon based compounds and other molecules may be explained in a similar way as methane. Take, for example, ethene (C2H4). Ethene has a double bond between the carbons.
For this molecule, carbon will sp2 hybridise, because one π (pi) bond is required for the double bond between the carbons, and only three σ bonds are formed per carbon atom. In sp2 hybridisation the 2s orbital is mixed with only two of the three available 2p orbitals:

forming a total of 3 sp2 orbitals with one p-orbital remaining. In ethylene (ethene) the two carbon atoms form a σ bond by overlapping two sp2 orbitals and each carbon atom forms two covalent bonds with hydrogen by s–sp2 overlap all with 120° angles. The π bond between the carbon atoms perpendicular to the molecular plane is formed by 2p–2p overlap. The hydrogen–carbon bonds are all of equal strength and length, which agrees with experimental data.
The amount of p-character is not restricted to integer values; i.e., hybridisations like sp2.5 are also readily described. In this case the geometries are somewhat distorted from the ideally hybridised picture. For example, as stated in Bent's rule, a bond tends to have higher p-character when directed toward a more electronegative substituent.
sp hybrids
 A schematic presentation of hybrid orbitals sp
A schematic presentation of hybrid orbitals spThe chemical bonding in compounds such as alkynes with triple bonds is explained by sp hybridization.

In this model, the 2s orbital mixes with only one of the three p-orbitals resulting in two sp orbitals and two remaining unchanged p orbitals. The chemical bonding in acetylene (ethyne) (C2H2) consists of sp–sp overlap between the two carbon atoms forming a σ bond and two additional π bonds formed by p–p overlap. Each carbon also bonds to hydrogen in a sigma s–sp overlap at 180° angles.
Hybridisation and molecule shape
Hybridisation, along with the VSEPR theory, helps to explain molecule shape:
- AX1 (e.g., LiH): no hybridisation; trivially linear shape
- AX2 (e.g., BeCl2): sp hybridisation; linear or digonal shape; bond angles are cos−1(−1) = 180°
- AX3 (e.g., BCl3): sp2 hybridisation; trigonal planar shape; bond angles are cos−1(−1/2) = 120°
- AX2E (e.g., GeF2): bent / V shape, < 120°
- AX4 (e.g., CCl4): sp3 hybridisation; tetrahedral shape; bond angles are cos−1(−1/3) ≈ 109.5°
- AX3E (e.g.,NH3): trigonal pyramidal, 107° (Note: The existence of a lone pair of electrons distorts bond angles slightly due to increased s-orbital character in the lone pair and increased p-orbital character in the orbitals used to make the bond pairs. It is not due to increased electron repulsion which is a very common misconception.[citation needed])
- AX5 (e.g., PCl5): sp3d hybridisation; trigonal bipyramidal shape
- AX6 (e.g., SF6): sp3d2 hybridisation; octahedral (or square bipyramidal) shape
This holds if there are no lone electron pairs on the central atom. If there are, they should be counted in the Xi number, but bond angles become smaller due to increased repulsion. For example, in water (H2O), the oxygen atom has two bonds with H and two lone electron pairs (as can be seen with the valence bond theory as well from the electronic configuration of oxygen), which means there are four such 'elements' on O. The model molecule is, then, AX2E2: sp3 hybridisation is utilized, and the electron arrangement of H2O is tetrahedral. This agrees with the experimentally-determined shape for water, a non-linear, bent structure, with a bond angle of 104.5 degrees (the two lone-pairs are not visible).
In general, for an atom with s and p orbitals forming hybrids hi and hj with included angle θ, the following holds: 1 + λiλj cos(θ) = 0. The p-to-s ratio for hybrid i is λi2, and for hybrid j it is λj2. In the special case of equivalent hybrids on the same atom, again with included angle θ, the equation reduces to just 1 + λ2 cos(θ) = 0. For example, BH3 has a trigonal planar geometry, three 120o bond angles, three equivalent hybrids about the boron atom, and thus 1 + λ2 cos(θ) = 0 becomes 1 + λ2 cos(120o) = 0, giving λ2 = 2 for the p-to-s ratio. In other words, sp2 hybrids, just as expected from the list above.
Explanation of the shape of water
Commonly, the hybridization of the oxygen in water is described as sp3 following the guidelines of VSEPR and the tetrahedral electron geometry it implies.[5] In order for this to be true, the two electron pairs would be in equal-energy, symmetrical, sp3 hybridised orbitals (two electron-pairs and two hydrogen atoms making the tetrahedron). However, molecular orbitals calculations give orbitals which reflect the symmetry of the molecule.[6] One of the two lone pairs is in a pure p-type orbital, with its electron density perpendicular to the H-O-H framework.[6] The other lone pair is an orbital that is close to an sp2-type orbital that is in the same plane as the H-O-H bonding. It is not a purely sp2-type orbital, but is extra rich in s-character.[6] Photoelectron spectra confirm the presence of two different energies for the nonbonded electrons.[7]
In contrast, the orbitals used to make the O-H bonds are close to sp2 hybrids, but are extra p-rich.[8] However, molecular orbital theory does not give two equivalent bonds, but two delocalised orbitals which are in-phase and out-of-phase combinations of the H-O bond orbitals.[6] It has been argued that it is this change in the mixing of the orbitals that is responsible for the compression of the H-O-H angle down to the experimental 104.5 degrees, not some change in the repulsion of electrons.[8] However, if the molecular orbitals are localised, leaving the total wave function unaltered, one obtains two equivalent lone pairs and two equivalent bonds.[9]
An accurate prediction of the bond angle requires however that polarisation d functions be added to the molecular orbital calculation.[10] Thus while VSEPR and its application to hybridisation predicts the correct atomic framework for water, it may do so for the wrong reason.
Controversy regarding d-orbital participation
Hybridisation theory has failed in a few aspects, notably in explaining the energy considerations for the involvement of d-orbitals in chemical bonding (See above for sp3d and sp3d2 hybridisation). This can be well-explained by means of an example. Consider, for instance, how the theory in question accounts for the bonding in phosphorus pentachloride (PCl5). The d-orbitals are large, comparatively distant from the nucleus and high in energy. Radial distances of orbitals from the nucleus seem to reveal that d-orbitals are far too high in energy to 'mix' with s- and p-orbitals. 3s – 0.47 , 3p – 0.55, 3d – 2.4 (in angstroms). Thus, at first glance, sp3d hybridisation seems improbable.
However, a closer examination of the factors that affect orbital size (and energy) reveals more. Formal charge on the central atom is one such factor, and it is obvious that the P atom in PCl5 carries quite a large partial positive charge. Thus the 3d orbital contracts in size to such an extent that hybridisation with s and p orbitals may occur. Further, note the cases in which d-orbital participation was proposed in hybridisation: SF6(sulfur hexafluoride), IF7, XeF6; in all these molecules, the central atom is surrounded by the highly electronegative fluorine atom, thus making hybridisation probable among s, p and d orbitals. A further study reveals that orbital size also depends on the number of electrons occupying it. And, even further, coupling of d orbital electrons also results in contraction, albeit to a lesser extent.
The molecular orbital theory, however, offers a clearer insight into the bonding in these molecules.
Hybridisation theory vs. MO theory
Hybridisation theory is an integral part of organic chemistry and in general discussed together with molecular orbital theory in advanced organic chemistry textbooks although for different reasons. One textbook notes that for drawing reaction mechanisms sometimes a classical bonding picture is needed with two atoms sharing two electrons.[11] It also comments that predicting bond angles in methane with MO theory is not straightforward. Another textbook treats hybridisation theory when explaining bonding in alkenes[12] and a third[13] uses MO theory to explain bonding in hydrogen but hybridisation theory for methane.
Although the language and pictures arising from hybridisation theory, more widely known as valence bond theory, remain widespread in synthetic organic chemistry, this qualitative view of bonding has been largely superseded by molecular orbital theory when a more detailed analysis is required. Advanced texts often stress that while hybrid orbital theory is still useful for problems requiring a rough approximation, it provides an incomplete picture that cannot account for many chemical phenomena.[14][15] One specific problem with hybridisation is that it incorrectly predicts the photoelectron spectra of many molecules, including such fundamental species such as methane and water. From a pedagogical perspective, hybridisation approach tends to over-emphasize localisation of bonding electrons and does not effectively embrace molecular symmetry as does MO theory.
Bonding orbitals formed from hybrid atomic orbitals may be considered as localized molecular orbitals, which can be formed from the delocalized orbitals of molecular orbital theory by an appropriate mathematical transformation. For molecules with a closed electron shell in the ground state, this transformation of the orbitals leaves the total many-electron wave function unchanged. The hybrid orbital description of the ground state is therefore equivalent to the delocalized orbital description for explaining the ground state total energy and electron density, as well as the molecular geometry which corresponds to the minimum value of the total energy.
There is no such equivalence, however, for ionized or excited states with open electron shells. Hybrid orbitals cannot therefore be used to interpret photoelectron spectra, which measure the energies of ionized states, identified with delocalized orbital energies using Koopmans' theorem. Nor can they be used to interpret UV-visible spectra which correspond to electronic transitions between delocalized orbitals.
See also
External links
- Covalent Bonds and Molecular Structure
- Hybridisation flash movie
- Hybridised orbital 3D preview progam in OpenGL
- Understanding Concepts: Molecular Orbitals
References
- ^ Gillespie, R.J. (2004), "Teaching molecular geometry with the VSEPR model", Journal of Chemical Education 81 (3): 298–304, doi:10.1021/ed081p298
- ^ Pauling, L. (1931), "The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules", Journal of the American Chemical Society 53 (4): 1367–1400, doi:10.1021/ja01355a027
- ^ McMurray, J. (1995). Chemistry Annotated Instructors Edition (4th ed.). Prentice Hall. p. 272. ISBN 0-13-140221-8
- ^ photo electron spectrum of methane 1 photo electron spectrum of methane 2
- ^ Petrucci R.H., Harwood W.S. and Herring F.G. "General Chemistry. Principles and Modern Applications" (Prentice-Hall 8th edn 2002) p.441
- ^ a b c d Levine I.N. “Quantum chemistry” (4th edn, Prentice-Hall) p.470-2
- ^ Levine p.475
- ^ a b Laing, Michael J. Chem. Educ. (1987) 64, 124–128 "No rabbit ears on water. The structure of the water molecule: What should we tell the students?"
- ^ Levine p.481-2
- ^ A.Szabo and N.S.Ostlund, "Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory" (MacMillan 1982) p.202 quotes calculated bond angles of 111.2° for a 4-31G basis set without d functions, and 105.5° for a 6-31G* basis set with d functions, compared to the expermimental 104.5°.
- ^ Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2001). Organic Chemistry (1st ed.). Oxford University Press. p. 105. ISBN 978-0-19-850346-0.
- ^ Organic Chemistry, Third Edition Marye Anne Fox James K. Whitesell 2003 ISBN 978-0-7637-3586-9
- ^ Organic Chemistry 3rd Ed. 2001 Paula Yurkanis Bruice ISBN 0-13-017858-6
- ^ G. L. Miessler and D. A. Tarr “Inorganic Chemistry” 3rd Ed, Pearson/Prentice Hall publisher, 2003. ISBN 0-13-035471-6.
- ^ Shriver, D. F.; Atkins, P. W.; Overton, T. L.; Rourke, J. P.; Weller, M. T.; Armstrong, F. A. “Inorganic Chemistry” W. H. Freeman, New York, 2006. ISBN 0-7167-4878-9.
Categories:- Chemical bonding
- Quantum chemistry
- Stereochemistry





{kind=link}
Wikimedia Foundation. 2010.