- Non-classical ion
-
Non-classical ions in organic chemistry are a special type of carbonium ions displaying delocalization of sigma bonds in 3-center-2-electron bonds of bridged systems.[1] The term non-classical ion was first used by John D. Roberts in 1951[2][3] in relation to the properties of cyclobutyl cations but the actual ions were first described by Saul Winstein in 1949 in order to explain the reactivity of certain norbornyl compounds.[4]
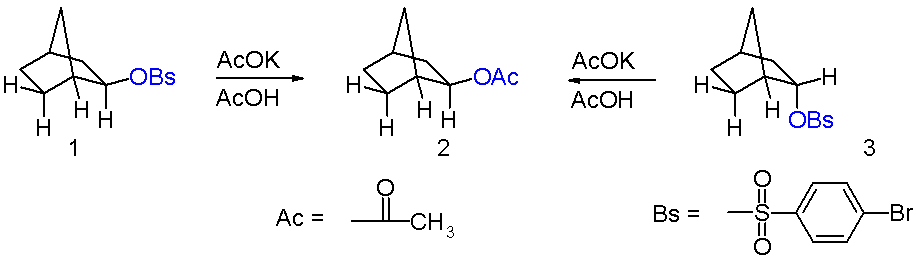
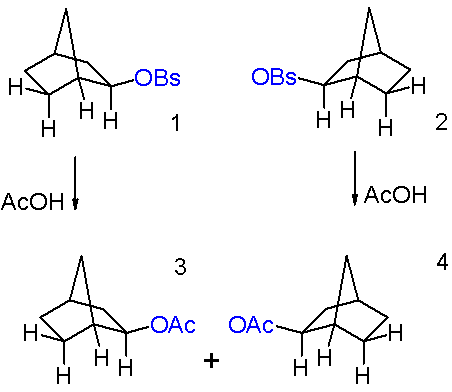
The compounds in question are exo-norbornyl brosylate 1 and its endo isomer 3 in scheme 1 and the reaction is solvolysis or acylation with the potassium salt of acetic acid in acetic acid. A key observation is that in this nucleophilic displacement both isomers give the same reaction product an exo-acetate 2. Also the reaction rate for the exo-reaction is 350 times the reaction rate for the endo reaction or a cyclohexyl control reaction
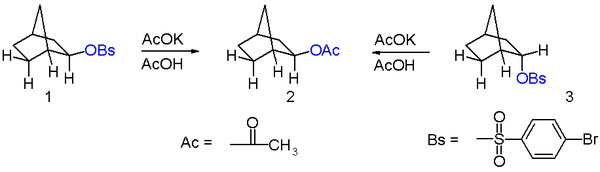
In a related experiment both enantiomers 1 and 2 of the exo-brosylate on solvolysis give the same racemic reaction product (scheme 2). The optical activity of the reaction disappears at the same reaction rate as that of the solvolysis.
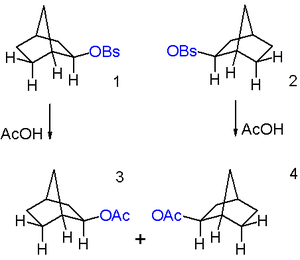
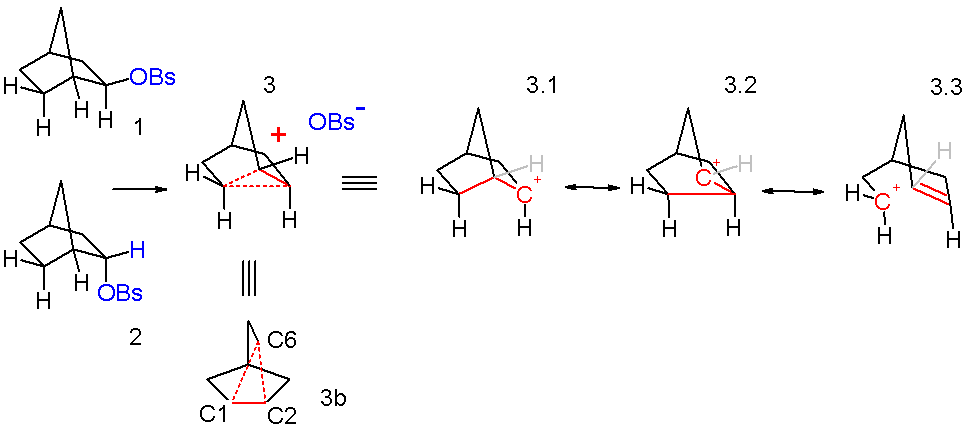
These observations are explained by invoking a non-classical ion 3 in scheme 3 as a reactive intermediate as the initial reaction product of both endo 1 and exo isomer 2. This ion is formed when sigma electrons in the C1-C6 bond assist by neighbouring group participation with the expulsion of the leaving group and now the positive charge residing on C1 is delocalized on C2 as well. The formation of the carbocation is the slow rate determining step. In this reaction step the exo leaving group is better positioned in relation to C1 than the endo leaving group and this explains the markedly difference in reactivity. The C2 carbon atom in the intermediate is pentavalent and therefore a carbonium ion. The ion is also symmetrical which is more obvious in the equivalent structure 3b. This symmetry explains the observed racemization. In classical resonance treatment the ion 3 can be regarded as a hybrid of resonance structures 3.1 through 3.3 with a full positive charge on C2, C6, and C7.
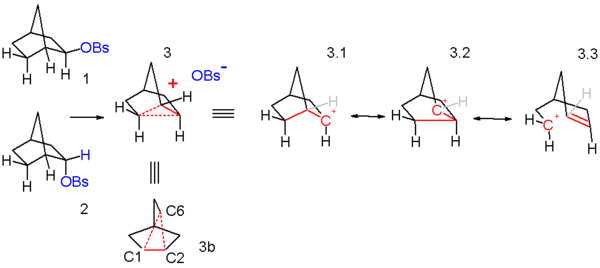
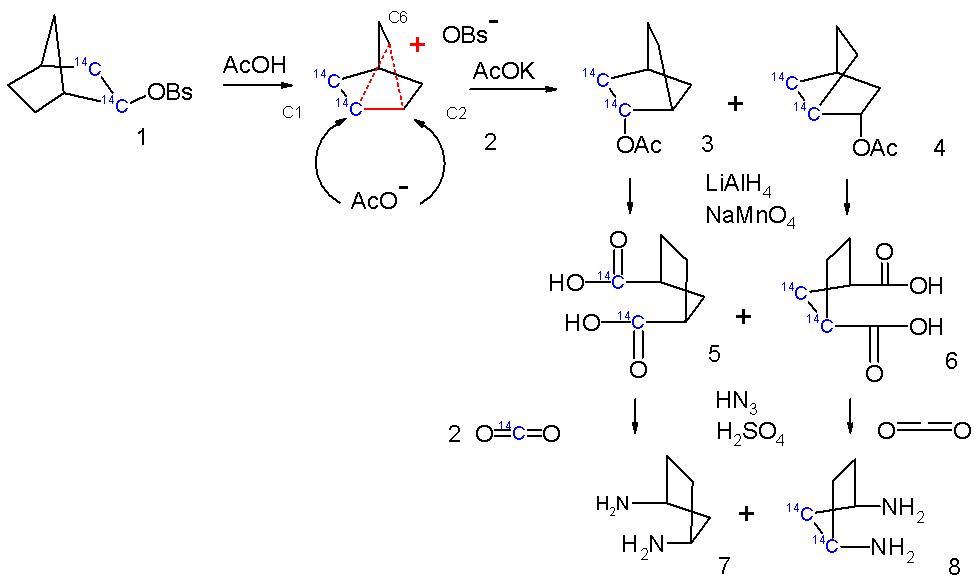
Further evidence for this model was obtained by tracer studies in which two carbon atoms in the norbornyl skeleton were replaced by the radioactive carbon 14 isotopes (Scheme 4)[citation needed]. The primary solvolyis reaction product was derivatized by reduction with lithium aluminium hydride and oxidation with sodium permanganate to the dicarboxylic acid followed by a Curtius rearrangement expelling carbon dioxide. When the acetyl anion is indeed able to attack both C1 and C2 positions of the symmetrical norbornyl cation 2 then 50% of the generated CO2 will contain 14C. The experimental value in this experiment was 40% and in order to account for the presence of 14C in other positions (a scrambling process) the nortricyclonium cation was postulated which is basically a face-capped norbornyl cation allowing hydride shifts.

In 1964 George A Olah began to produce direct evidence for the norbornyl cation when he subjected the norbornyl antimony chloropentafluoride salt obtained by reaction of exo-2-chloro-norbornane with antimony pentafluoride to NMR analysis.[5] The room temperature NMR spectrum was a single broad peak due to the presence of hydride shifts but these could be partially frozen when cooled to −60 °C in liquid sulfur dioxide. It was also found that the norbornyl cation could also be generated from norbornanes with chlorine substituent at the bridging position or bridgehead positions and from reaction of norbornene with HSbF6 thereby confirming the presence of the rearrangements taking place in the 14C scrambling process.
Solid-state NMR analysis was possible at temperatures as low as 5 kelvins [6] at which temperature all positions are assumed to be frozen. One of the two signals visible in the spectrum corresponded to the identical C1 and C2 carbon atoms.
See also
- organic chemistry
- reactive intermediate
- carbocation
External links
References
- ^ Moss, Robert A. / Platz, Matthew S. / Jones, Maitland (eds.) Reactive Intermediate Chemistry ISBN 0-471-23324-2 - John Wiley & Sons
- ^ J. D. Roberts and R. H. Mazur (1951). "The nature of the intermediate in carbonium ion-type interconversion reactions of cyclobutyl, cyclopropylcarbinyl and allylcarbinyl derivatives". J. Am. Chem. Soc. 73 (7): 3542–3543. doi:10.1021/ja01151a550.
- ^ J. D. Roberts and C. C. Lee (1951). "The nature of the intermediate in the solvolysis of norbornyl derivatives". J. Am. Chem. Soc. 73 (10): 5009–5010. doi:10.1021/ja01154a555.
- ^ Saul Winstein and D. S. Trifan (1949). "The structure of the bicyclo[2,2,1]2-heptyl (norbornyl) carbonium ion". J. Am. Chem. Soc. 71 (8): 2953–2953. doi:10.1021/ja01176a536.
- ^ P. v. R. Schleyer, W. E. Watts, R. C. Fort, M. B. Comisarow and G. A. Olah (1964). "Stable Carbonium Ions. X.1 Direct Nuclear Magnetic Resonance Observation of the 2-Norbornyl Cation". J. Am. Chem. Soc. 86 (24): 5679–5680. doi:10.1021/ja01078a056.
- ^ C. S. Yannoni, V. Macho and P. C. Myhre (1982). "Resolved 13 C NMR spectra of carbonium ions at cryogenic temperatures. The norbornyl cation at 5 K". J. Am. Chem. Soc. 104 (25): 7380–7381. doi:10.1021/ja00389a108.
Categories:- Reactive intermediates
- Physical organic chemistry
Wikimedia Foundation. 2010.