- Crystallographic database
-
A crystallographic database is a database specifically designed to store information about crystals and crystal structures. Crystals are solids having, in all three dimensions of space, a regularly repeating arrangement of atoms, ions, or molecules. They are characterized by symmetry, morphology, and directionally dependent physical properties. A crystal structure describes the arrangement of atoms, ions, or molecules in a crystal.
Crystal structures of crystalline material are typically determined from X-ray or neutron single-crystal diffraction data and stored in crystal structure databases. They are routinely identified by comparing reflection intensities and lattice spacings from X-ray powder diffraction data with entries in powder-diffraction fingerprinting databases.
Crystal structures of nanometer sized crystalline samples can be determined via structure factor amplitude information from single-crystal electron diffraction data or structure factor amplitude and phase angle information from Fourier transforms of HRTEM images of crystallites. They are stored in crystal structure databases specializing in nanocrystals and can be identified by comparing zone axis subsets in lattice-fringe fingerprint plots with entries in a lattice-fringe fingerprinting database.
Crystallographic databases can be categorized as crystallographic information from supersets or subsets of inorganics, metals/alloys, organics, and biological macromolecules. They differ in access and usage rights and offer varying degrees of search and analysis capacity. Many provide structure visualization capabilities. They can be browser based or installed locally. Newer versions are built on the relational database model and support the Crystallographic Information File (CIF) as a universal data exchange format.
Contents
Overview
Crystallographic data are primarily extracted from published scientific articles and supplementary material. Newer versions of crystallographic databases are built on the relational database model, which enables efficient cross-referencing of tables. Cross-referencing serves to derive additional data or enhance the search capacity of the database.
Data exchange among crystallographic databases, structure visualization software, and structure refinement programs has been facilitated by the emergence of the Crystallographic Information File (CIF) format. The CIF format is the standard text file format for the exchange and archiving of crystallographic data.[1] It was adopted by the International Union of Crystallography (IUCr), who also provides full specifications of the format.[2] It is supported by all major crystallographic databases.
The increasing automation of the crystal structure determination process has resulted in ever higher publishing rates of new crystal structures and, consequentially, new publishing models. Minimalistic articles contain only crystal structure tables, structure images, and, possibly, abstract-like structure description. They tend to be published in author-financed or subsidized open-access journals. Acta Crystallographica Section E and Zeitschrift für Kristallographie belong in this category. More elaborate contributions may go to traditional subscriber-financed journals. Hybrid journals, on the other hand, embed individual author-financed open-access articles among subscriber-financed ones. Publishers may also make scientific articles available online, as Portable Document Format (PDF) files.
Crystal structure data in CIF format are linked to scientific articles as supplementary material. CIFs may be accessible directly from the publisher’s website, crystallographic databases, or both. In recent years, many publishers of crystallographic journals have come to interpret CIFs as formatted versions of open data, i.e. representing non-copyrightable facts, and therefore tend to make them freely available online, independent of the accessibility status of linked scientific articles.
Trends
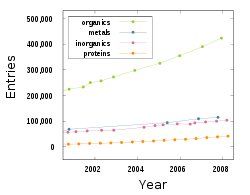 Trends of crystal structures in databases over the last decade.[3]
Trends of crystal structures in databases over the last decade.[3]
As of 2008, more than 700,000 crystal structures have been published and stored in crystal structure databases. The publishing rate has reached more than 50,000 crystal structures per year. These numbers refer to published and republished crystal structures from experimental data. Crystal structures are republished owing to corrections for symmetry errors, improvements of lattice and atomic parameters, and differences in diffraction technique or experimental conditions.
Crystal structures are typically categorized as minerals, metals-alloys,[4] inorganics,[5] organics,[6] nucleic acids,[7] and biological macromolecules.[8] Individual crystal structure databases cater for users in specific chemical, molecular-biological, or related disciplines by covering super- or subsets of these categories. Minerals are a subset of mostly inorganic compounds. The category ‘metals-alloys’ covers metals, alloys, and intermetallics. Metals-alloys and inorganics can be merged into ‘non-organics’. Organic compounds and biological macromolecules are separated according to molecular size. Organic salts, organometallics, and metalloproteins tend to be attributed to organics or biological macromolecules, respectively. Nucleic acids are a subset of biological macromolecules.
Comprehensiveness can refer to the number of entries in a database. On those terms, a crystal structure database can be regarded as comprehensive, if it contains a gapless collection of all (re-)published crystal structures in the category of interest and is updated frequently. Searching for structures in such a database can replace more time-consuming scanning of the open literature. Access to crystal structure databases differs widely. It can be divided into reading and writing access. Reading access rights (search, download) affect the number and range of users. Restricted reading access is often coupled with restricted usage rights. Writing access rights (upload, edit, delete), on the other hand, determine the number and range of contributors to the database. Restricted writing access is often coupled with high data integrity.
In terms of user numbers and daily access rates, comprehensive and thoroughly vetted open-access crystal structure databases naturally surpass comparable databases with more restricted access and usage rights. Independent of comprehensiveness, open-access crystal structure databases can spawn open-source software projects, such as search-analysis tools, visualization software, and derivative databases. Scientific progress can be slowed down by restricting access or usage rights as well as limiting comprehensiveness or data integrity. Restricted access or usage rights are commonly associated with commercial crystal structure databases. Lack of comprehensiveness or data integrity, on the other hand, are associated with some of the open-access crystal structure databases. Apart from that, several crystal structure databases are freely available for primarily educational purposes, in particular mineralogical databases.
Crystallographic databases can specialize in crystal structures, crystal phase identification, crystallization,[9] crystal morphology, or various physical properties. More integrative databases combine several categories of compounds or specializations.[10] Structures of incommensurate phases, nanocrystals, thin films on substrates,[11] and predicted crystal structures are collected in tailored special structure databases.
Search
Search capacities of crystallographic databases differ widely. Basic functionality comprises search by keywords, physical properties, and chemical elements. Of particular importance is search by compound name and lattice parameters. Very useful are search options that allow the use of wildcard characters and logical connectives in search strings. If supported, the scope of the search can be constrained by the exclusion of certain chemical elements.
More sophisticated algorithms depend on the material type covered. Organic compounds might be searched for on the basis of certain molecular fragments. Inorganic compounds, on the other hand, might be of interest with regard to a certain type of coordination geometry. More advanced algorithms deal with conformation analysis (organics), supramolecular chemistry (organics), interpolyhedral connectivity (‘non-organics’) and higher-order molecular structures (biological macromolecules). Search algorithms used for a more complex analysis of physical properties, e.g. phase transitions or structure-property relationships, might apply group-theoretical concepts.
Modern versions of crystallographic databases are based on the relational database model. Communication with the database usually happens via a dialect of the Structured Query Language (SQL). Web-based databases typically process the search algorithm on the server interpreting supported scripting elements, while desktop-based databases run locally installed and usually precompiled search engines.
Crystal phase identification
Crystalline material may be divided into single crystals, twin crystals, polycrystals, and crystal powder. In a single crystal, the arrangement of atoms, ions, or molecules is defined by a single crystal structure in one orientation. Twin crystals, on the other hand, consist of single-crystalline twin domains, which are aligned by twin laws and separated by domain walls.
Polycrystals are made of a large number of small single crystals, or crystallites, held together by thin layers of amorphous solid. Crystal powder is obtained by grinding crystals, resulting in powder particles, made up of one or more crystallites. Both polycrystals and crystal powder consist of many crystallites with varying orientation.
Crystal phases are defined as regions with the same crystal structure, irrespective of orientation or twinning. Single and twinned crystalline specimens therefore constitute individual crystal phases. Polycrystalline or crystal powder samples may consist of more than one crystal phase. Such a phase comprises all the crystallites in the sample with the same crystal structure.
Crystal phases can be identified by successfully matching suitable crystallographic parameters with their counterparts in database entries. Prior knowledge of the chemical composition of the crystal phase can be used to reduce the number of database entries to a small selection of candidate structures and thus simplify the crystal phase identification process considerably.
Powder diffraction fingerprinting (1D)
Applying standard diffraction techniques to crystal powders or polycrystals is tantamount to collapsing the 3D reciprocal space, as obtained via single-crystal diffraction, onto a 1D axis. The resulting partial-to-total overlap of symmetry-independent reflections renders the structure determination process more difficult, if not impossible.
Powder diffraction data can be plotted as diffracted intensity (I) versus reciprocal lattice spacing (1/d). Reflection positions and intensities of known crystal phases, mostly from X-ray diffraction data, are stored, as d-I data pairs, in the Powder Diffraction File (PDF) database. The list of d-I data pairs is highly characteristic of a crystal phase and, thus, suitable for the identification, also called ‘fingerprinting’, of crystal phases.[12]
Search-match algorithms compare selected test reflections of an unknown crystal phase with entries in the database. Intensity-driven algorithms utilize the three most intense lines (so-called ‘Hanawalt search’), while d-spacing-driven algorithms are based on the eight to ten largest d-spacings (so-called ‘Fink search’).[13]
X-ray powder diffraction fingerprinting has become the standard tool for the identification of single or multiple crystal phases and is widely used in such fields as metallurgy, mineralogy, forensic science, archeology, condensed matter physics, and the biological and pharmaceutical sciences.
Lattice-fringe fingerprinting (2D)
Main article: lattice-fringe fingerprintingPowder diffraction patterns of very small single crystals, or crystallites, are subject to size-dependent peak broadening, which, below a certain size, renders powder diffraction fingerprinting useless. In this case, peak resolution is only possible in 3D reciprocal space, i.e. by applying single-crystal electron diffraction techniques.
High-Resolution Transmission Electron Microscopy (HRTEM) provides images and diffraction patterns of nanometer sized crystallites. Fourier transforms of HRTEM images and electron diffraction patterns both supply information about the projected reciprocal lattice geometry for a certain crystal orientation, where the projection axis coincides with the optical axis of the microscope.
Projected lattice geometries can be represented by so-called ‘lattice-fringe fingerprint plots’ (LFFPs), also called angular covariance plots.[14] The horizontal axis of such a plot is given in reciprocal lattice length and is limited by the point resolution of the microscope. The vertical axis is defined as acute angle between Fourier transformed lattice fringes or electron diffraction spots. A 2D data point is defined by the length of a reciprocal lattice vector and its (acute) angle with another reciprocal lattice vector. Sets of 2D data points that obey Weiss’s zone law are subsets of the entirety of data points in an LFFP. A suitable search-match algorithm using LFFPs, therefore, tries to find matching zone axis subsets in the database. It is, essentially, a variant of a lattice matching algorithm.[15]
The performance of search-match procedures utilizing LFFPs, also called ‘lattice-fringe fingerprinting’, can be sped up by precalculating and storing full LFFPs of all entries, assuming either kinematic or dynamic scattering and a given point resolution of the microscope. The number of possible entries can be narrowed down on the basis of chemical compound information.[16]
In the case of electron diffraction patterns, structure factor amplitudes can be used, in a later step, to further discern among a selection of candidate structures (so-called 'structure factor fingerprinting'). Structure factor amplitudes from electron diffraction data are far less reliable than their counterparts from X-ray single-crystal and powder diffraction data. Existing precession electron diffraction techniques greatly improve the quality of structure factor amplitudes, increase their number and, thus, make structure factor amplitude information much more useful for the fingerprinting process.[17]
Fourier transforms of HRTEM images, on the other hand, supply information not only about the projected reciprocal lattice geometry and structure factor amplitudes, but also structure factor phase angles. After crystallographic image processing,[18] structure factor phase angles are far more reliable than structure factor amplitudes. Further discernment of candidate structures is then mainly based on structure factor phase angles and, to a lesser extent, structure factor amplitudes (so-called 'structure factor fingerprinting').[19][20]
Morphological fingerprinting (3D)
The Generalized Steno Law[21] states that the interfacial angles between identical faces of any single crystal of the same material are, by nature, restricted to the same value.[22] This offers the opportunity to fingerprint crystalline materials on the basis of optical goniometry, which is also known as crystallometry.[23] In order to employ this technique successfully, one must consider the observed point group symmetry of the measured faces and creatively apply the rule that "crystal morphologies are often combinations of simple (i.e. low multiplicity) forms where the individual faces have the lowest possible Miller indices for any given zone axis". This shall ensure that the correct indexing of the crystal faces is obtained for any single crystal.
It is in many cases possible to derive the ratios of the crystal axes for crystals with low symmetry from optical goniometry with high accuracy and precision and to identify a crystalline material on their basis alone employing databases such as 'Crystal Data'.[24] Provided that the crystal faces have been correctly indexed and the interfacial angles were measured to better than a few fractions of a tenth of a degree, a crystalline material can be identified quite unambiguously on the basis of angle comparisons to two rather comprehensive databases: the 'Bestimmungstabellen für Kristalle (Определитель Кристаллов)'[25] and the 'Barker Index of Crystals'.[26]
Since Steno’s Law can be further generalized for a single crystal of any material to include the angles between either all identically indexed net planes (i.e. vectors of the reciprocal lattice, also known as 'potential reflections in diffraction experiments') or all identically indexed lattice directions (i.e. vectors of the direct lattice, also known as zone axes), opportunities exist for morphological fingerprinting of nanocrystals in the transmission electron microscope (TEM) by means of transmission electron goniometry.[27]
The specimen goniometer of a TEM is thereby employed analogously to the goniometer head of an optical goniometer. The optical axis of the TEM is then analogous to the reference direction of an optical goniometer. While in optical goniometry net-plane normals (reciprocal lattice vectors) need to be successively aligned parallel to the reference direction of an optical goniometer in order to derive measurements of interfacial angles, the corresponding alignment needs to be done for zone axes (direct lattice vector) in transmission electron goniometry. (Note that such alignments are by their nature quite trivial for nanocrystals in a TEM after the microscope has been aligned by standard procedures.)
Since transmission electron goniometry is based on Bragg’s Law for the transmission (Laue) case (diffraction of electron waves), interzonal angles (i.e. angles between lattice directions) can be measured by a procedure that is analogous to the measurement of interfacial angles in an optical goniometer on the basis of Snell’s Law, i.e. the reflection of light. The complements to interfacial angles of external crystal faces can, on the other hand, be directly measured from a zone-axis diffraction pattern or from the Fourier transform of a high resolution TEM image that shows crossed lattice fringes.
Lattice matching (3D)
Lattice parameters of unknown crystal phases can be obtained from X-ray, neutron, or electron diffraction data. Single-crystal diffraction experiments supply orientation matrices, from which lattice parameters can be deduced. Alternatively, lattice parameters can be obtained from powder or polycrystal diffraction data via profile fitting without structural model (so-called 'Le Bail method').
Arbitrarily defined unit cells can be transformed to a standard setting and, from there, further reduced to a primitive smallest cell. Sophisticated algorithms compare such reduced cells with corresponding database entries. More powerful algorithms also consider derivative super- and subcells. The lattice-matching process can be further sped up by precalculating and storing reduced cells for all entries. The algorithm searches for matches within a certain range of the lattice parameters. More accurate lattice parameters allow a narrower range and, thus, a better match.[28]
Lattice matching is useful in identifying crystal phases in the early stages of single-crystal diffraction experiments and, thus, avoiding unnecessary full data collection and structure determination procedures for already known crystal structures. The method is particularly important for single-crystalline samples that need to be preserved. If, on the other hand, some or all of the crystalline sample material can be ground, powder diffraction fingerprinting is usually the better option for crystal phase identification, provided that the peak resolution is good enough. However, lattice matching algorithms are still better at treating derivative super- and subcells.
Visualization
Newer versions of crystal structure databases integrate the visualization of crystal and molecular structures. Specialized or integrative crystallographic databases may provide morphology or tensor visualization output.
Crystal structures
The crystal structure describes the three-dimensional periodic arrangement of atoms, ions, or molecules in a crystal. The unit cell represents the simplest repeating unit of the crystal structure. It is a parallelepiped containing a certain spatial arrangement of atoms, ions, molecules, or molecular fragments. From the unit cell the crystal structure can be fully reconstructed via translations.
The visualization of a crystal structure can be reduced to the arrangement of atoms, ions, or molecules in the unit cell, with or without cell outlines. Structure elements extending beyond single unit cells, such as isolated molecular or polyhedral units as well as chain, net, or framework structures, can often be better understood by extending the structure representation into adjacent cells.
The space group of a crystal is a mathematical description of the symmetry inherent in the structure. The motif of the crystal structure is given by the asymmetric unit, a minimal subset of the unit cell contents. The unit cell contents can be fully reconstructed via the symmetry operations of the space group on the asymmetric unit. Visualization interfaces usually allow for switching between asymmetric unit and full structure representations.
Bonds between atoms or ions can be identified by characteristic short distances between them. They can be classified as covalent, ionic, hydrogen, or other bonds including hybrid forms. Bond angles can be deduced from the bond vectors in groups of atoms or ions. Bond distances and angles can be made available to the user in tabular form or interactively, by selecting pairs or groups of atoms or ions. In ball-and-stick models of crystal structures, balls represent atoms and sticks represent bonds.
Since organic chemists are particularly interested in molecular structures, it might be useful to be able to single out individual molecular units interactively from the drawing. Organic molecular units need to be given both as 2D structural formulae and full 3D molecular structures.[29] Molecules on special-symmetry positions need to be reconstructed from the asymmetric unit. Protein crystallographers are interested in molecular structures of biological macromolecules, so that provisions need to be made to be able to represent molecular subunits as helices, sheets, or coils, respectively.
Crystal structure visualization can be integrated into a crystallographic database. Alternatively, the crystal structure data are exchanged between the database and the visualization software, preferably using the CIF format.[30] Web-based crystallographic databases can integrate crystal structure visualization capability.[31] Depending on the complexity of the structure, lighting, and 3D effects, crystal structure visualization can require a significant amount of processing power, which is why the actual visualization is typically run on the client.
Currently, web-integrated crystal structure visualization is based on Java applets from open-source projects such as Jmol.[32] Web-integrated crystal structure visualization is tailored for examining crystal structures in web browsers, often supporting wide color spectra (up to 32 bit) and window size adaptation. However, web-generated crystal structure images are not always suitable for publishing due to issues such as resolution depth, color choice, grayscale contrast, or labeling (positioning, font type, font size).[33]
Morphology and physical properties
Mineralogists, in particular, are interested in morphological appearances of individual crystals, as defined by the actually formed crystal faces (tracht) and their relative sizes (habit). More advanced visualization capabilities allow for displaying surface characteristics, imperfections inside the crystal, lighting (reflection, shadow, and translucency), and 3D effects (interactive rotatability, perspective, and stereo viewing).[34][35]
Crystal physicists, in particular, are interested in anisotropic physical properties of crystals. The directional dependence of a crystal's physical property is described by a 3D tensor and depends on the orientation of the crystal. Tensor shapes are more palpable by adding lighting effects (reflection and shadow). 2D sections of interest are selected for display by rotating the tensor interactively around one or more axes.[36]
Crystal morphology or physical property data can be stored in specialized databases or added to more comprehensive crystal structure databases. The Crystal Morphology Database (CMD) is an example for a web-based crystal morphology database with integrated visualization capabilities.
See also
References
- ^ Brown I. David, McMahon Brian (2002). "CIF: The Computer Language of Crystallography". Acta Crystallographica Section B 58 (3): 317–324. doi:10.1107/S0108768102003464.
- ^ Sydney Hall and Brian McMahon (2005). International Tables for Crystallography, Volume G. Springer. ISBN 978-1-4020-3138-0.
- ^ Sources:
- ^ White Peter S., Rodgers John R., Yvon Le Page (2002). "CRYSTMET: A Database of the Structures and Powder Patterns of Metals and Intermetallics". Acta Crystallographica Section B 58 (3): 343–348. doi:10.1107/S0108768102002902.
- ^ Belsky Alec, Hellenbrandt Mariette, Lynn Vicky, Karen , Luksch Peter (2002). "New Developments in the Inorganic Crystal Structure Database (ICSD): Accessibility in Support of Materials Research and Design". Acta Crystallographica Section B 58 (3): 364–369. doi:10.1107/S0108768102006948.
- ^ Allen Frank H (2002). "The Cambridge Structural Database: a Quarter of a Million Crystal Structures and Rising". Acta Crystallographica Section B 58 (3): 380–388. doi:10.1107/S0108768102003890.
- ^ Berman Helen M., Westbrook John, Feng Zukang, Iype Lisa, Schneider Bohdan, Zardecki Christine (2002). "The Nucleic Acid Database". Acta Crystallographica Section D 58: 889–898.
- ^ Berman Helen M., Battistuz Tammy, Bhat T. N., Bluhm Wolfgang F., Bourne Philip E., Burkhardt Kyle, Feng Zukang, Gilliland Gary L., Iype Lisa et al. (2002). "The Protein Data Bank". Acta Crystallographica Section D 58: 899–907.
- ^ Gilliland Gary L., Tung Michael, Ladner Jane E. (2002). "The Biological Macromolecule Crystallization Database: Crystallization Procedures and Strategies". Acta Crystallographica Section D 58: 916–920.
- ^ Pierre Villars, N. Onodera, and Shuichi Iwata, "The Linus Pauling File and its Application to Materials Design", Journal of Alloys and Compounds, 279(1) (1998) pp. 1-7.
- ^ Michel A. Van Hove, Klaus Hermann, and Philip R. Watson, "The NIST Surface Structure Database - SSD Version 4", Acta Crystallographica Section B, 58 (2002) pp. 338-342.
- ^ Faber John, Fawcett Tim (2002). "The Powder Diffraction File: Present and Future". Acta Crystallographica Section B 58 (3): 325–332. doi:10.1107/S0108768102003312.
- ^ John Faber, Tim Fawcett, "New Powder Diffraction File (PDF-4) in Relational Database Format: Advantages and Data-Mining Capabilities", Acta Crystallographica Section B, 58 (2002) pp. 333-337.
- ^ Philip Fraundorf, Wentao Quin, Peter Moeck, and Eric Mandell (2005). "Making Sense of Nanocrystal Lattice Fringes". Journal of Applied Physics 98 (11): pp. 114308–1–114308–10. arXiv:cond-mat/0212281. Bibcode 2005JAP....98k4308F. doi:10.1063/1.2135414.
- ^ Peter Moeck (2008). Structural Identification of Cubic Iron-Oxide Nanocrystal Mixtures: X-ray Powder Diffraction Versus Quasi-Kinematic Transmission Electron Microscopy", arXiv:0804.0063]]. 0804. pp. 63. arXiv:0804.0063. Bibcode 2008arXiv0804.0063M.
- ^ Ruben Bjorge (2007.). Lattice-Fringe Fingerprinting: Structural Identification of Nanocrystals Employing High-Resolution Transmission Electron Microscopy", Master of Science Thesis, Portland State University. http://www.scientificjournals.org/journals2007/articles/1235.pdf.
- ^ Xiadong Zou and Sven Hovmöller (2008). "Electron Crystallography: Imaging and Single-Crystal Diffraction from Powders". Acta Crystallographica Section A 64: 149–169.. doi:10.1107/S0108767307060084. http://journals.iucr.org/a/issues/2008/01/00/issconts.html.
- ^ Hovmöller S (1992). "CRISP: Crystallographic Image Processing on a Personal Computer". Ultramicroscopy 41: 121–135. doi:10.1016/0304-3991(92)90102-P.
- ^ Peter Moeck and Philip Fraundorf (2007). "Structural fingerprinting in the transmission electron microscope: overview and opportunities to implement enhanced strategies for nanocrystal identification". Zeitschrift für Kristallographie 222 (11): 634. doi:10.1524/zkri.2007.222.11.634. http://www.atypon-link.com/OLD/doi/pdf/10.1524/zkri.2007.222.11.634.
- ^ [1], "Database Supported Nanocrystal Structure Identification by Lattice-Fringe Fingerprinting with Structure Factor Extraction"
- ^ N. Steno, De solido intra solidum naturaliter contento dissertations prodromus, Florence 1669; translated by Karl Mieleitner, Vorläufer einer Dissertation über feste Körper, die innerhalb anderer fester Körper von Natur aus eingeschlossen sind, Leipzig, 1923.
- ^ J. B. L. Romé de l’Isle, Cristallographie, ou description des formes propres à tous les corps du règne minéral (4 vol. Paris, 1783).
- ^ P. Terpstra and L. W. Codd (1961). Crystallometry. New York:Academic Press.
- ^ J. D. H. Donnay, C. Donnay, E. G. Cox, O. Kennard, M. V. King, Crystal Data, Monography 5, American Crystallographic Association, Washington, William and Heintz, 1963.
- ^ A. K. Boldyrew and W. W. Doliwo-Dobrowolsky, Bestimmungstabellen für Kristalle (Определитель Кристаллов), Vol. I, Part 1, Einleitung, Tetragyrische Syngonie; W.W. Doliwo-Dobrowolsky and G.P. Preobraschensky, Vol. I, Part 2, Trigyrische and Hexagyrische Syngonien allgemeine Ergänzungen zu den mittleren Syngonien, Zentrales Wissenschaftliches Institut der Geologie und Schürfung, Leningrad and Moscow, 1937 and 1939.
- ^ M. W. Porter and R. C. Spiller, The Barker Index of Crystals, Vol. I and II, W. Heffer and Sons, Cambridge, 1951 and 1956; M. W. Porter and W. L. Codd, The Barker Index of Crystals, Vol. III, W. Heffer and Sons, Cambridge, 1964.
- ^ Peter Moeck and Philip Fraundorf (2006-09-14). "Transmission Electron Goniometry and its Relation to Electron Tomography for Materials Science Applications". Eprint arXiv:cond-mat/0611345 2: 11345. arXiv:cond-mat/0611345. Bibcode 2006cond.mat.11345M.
- ^ Frank H. Allen, Günther Bergerhoff, Rolf Sievers (1987). Crystallographic Databases. Chester: IUCr.
- ^ Bruno Ian J., Cole Jason C., Edgington Paul R., Kessler Magnus, Macrae Clare F., McCabe Patrick, Pearson Jonathan, Taylor Robin (2002). "New software for searching the Cambridge Structural Database and visualizing crystal structures". Acta Crystallographica Section B 58 (3): 389–397. doi:10.1107/S0108768102003324.
- ^ Allen Frank H., Motherwell W. D. Samuel (2002). "Applications of the Cambridge Structural Database in Organic Chemistry and Crystal Chemistry". Acta Crystallographica Section B 58 (3): 407–422. doi:10.1107/S0108768102004895.
- ^ Peter Moeck, Ondřej Čertík, Girish Upreti, Björn Seipel, Morgan Harvey, William Garrick, Philip Fraundorf, "Crystal Structure Visualizations in three Dimensions with Support from the Open Access Nano-Crystallography Database", Journal of Materials Education, 28(1) (2006) pp. 83-90.
- ^ Marion E. Cass, Henry S. Rzepa, David R. Rzepa, Charlotte K. Williams, "The Use of the Free, Open-Source Program Jmol to Generate an Interactive Web Site to Teach Molecular Symmetry", Journal of Chemical Education, 82(11) (2005) pp. 1736-1740.
- ^ Angel Herraez, "Biomolecules in the Computer - Jmol to the Rescue", Biochemistry and Molecular Biology Education 34(4) (2006) pp. 255-261.
- ^ Werner Kaminsky, "From CIF to Virtual Morphology Using the WinXMorph Program", Journal of Applied Crystallography, 40(2) (2007) pp. 382-385.
- ^ Werner Kaminsky, "WinXMorph: a Computer Program to Draw Crystal Morphology, Growth Sectors and Cross Sections with Export Files in VRML V2.0 UTF8-Virtual Reality Format", Journal of Applied Crystallography, 38(3) (2005) pp. 566-567.
- ^ Kaminsky Werner (2000). "Wintensor: Ein WIN95/98/NT Programm zum Darstellen tensorieller Eigenschaften". Zeitschrift für Kristallographie Supplement 17: 51.
External links
Crystal structures
- American Mineralogist Crystal Structure Database (AMCSD) (contents: crystal structures of minerals, access: free, size: medium)
- Cambridge Structural Database (CSD) (contents: crystal structures of organics and metal-organics, access: restricted, size: large)
- Crystal Lattice Structures (contents: a selection of common crystal structures, access: free, size: small)
- Crystallography Open Database (COD) (contents: crystal structures of organics, metalorganics, minerals, inorganics, metals, alloys, and intermetallics, access: free, size: medium - large)
- Database of Zeolite Structures (contents: crystal structures of zeolites, access: free, size: small)
- Incommensurate Structures Database (contents: incommensurate structures, access: free, size: small)
- Inorganic Crystal Structure Database (ICSD) (contents: crystal structures of minerals and inorganics, access: restricted, size: large)
- Metals Structure Database (CRYSTMET) (contents: crystal structures of metals, alloys, and intermetallics, access: restricted, size: large)
- Mineralogy Database (contents: crystal structures of minerals, access: free, size: medium)
- MinCryst (contents: crystal structures of minerals, access: free, size: medium)
- Nano-Crystallography Database (NCD) (contents: crystal structures of nanometer sized crystallites, access: free, size: small)
- NIST Structural Database NIST Structural Database (contents: crystal structures of metals, alloys, and intermetallics, access: restricted, size: large)
- NIST Surface Structure Database (contents: surface and interface structures, access: restricted, size: small-medium)
- Nucleic Acid Database (contents: crystal and molecular structures of nucleic acids, access: free, size: medium)
- Pearson's Crystal Data (contents: crystal structures of organics, metalorganics, minerals, inorganics, metals, alloys, and intermetallics, access: restricted, size: large)
- Worldwide Protein Data Bank (PDB) (contents: crystal and molecular structures of biological macromolecules, access: free, size: medium-large)
- Wiki Crystallography Database (WCD) (contents: crystal structures of organics, metalorganics, minerals, inorganics, metals, alloys, and intermetallics, access: free, size: medium)
Crystal phase identification
- Match! (method: powder diffraction fingerprinting)
- Nano-Crystallography Database (NCD) (method: lattice-fringe fingerprinting)
- NIST Crystal Data (method: lattice matching)
- Powder Diffraction File (PDF) (method: powder diffraction fingerprinting)
- Wiki Crystallography Database (WCD) (method: lattice-fringe fingerprinting)
Specialized databases
- Biological Macromolecule Crystallization Database (BMCD) (specialization: crystallization of biological macromolecules, access: free, size: medium)
- Crystal Morphology Database (CMD) (specialization: morphology of crystals, access: free, size: small)
- Database of Zeolite Structures (specialization: crystal structures of zeolites, access: free, size: small)
- Incommensurate Structures Database (specialization: incommensurate structures, access: free, size: small)
- Marseille Protein Crystallization Database (MPCD) (specialization: crystallization of biological macromolecules, access: free, size: medium)
- Nano-Crystallography Database (NCD) (specialization: crystal structures of nanometer sized crystallites, access: free, size: small)
- NIST Surface Structure Database (specialization: surface and interface structures, access: restricted, size: small-medium)
- Linus Pauling File (LPF) (specialization: physical properties of metals, alloys, intermetallics, and inorganics, access: free, size: medium-large)
- Predicted Crystallography Open Database (PCOD) (spezialization: predicted crystal structures of organics, metal-organics, metals, alloys, intermetallics, and inorganics, access: free, size: large)
Categories:- Crystallography
- Databases
- Physical chemistry

Wikimedia Foundation. 2010.