- Gene expression profiling
-
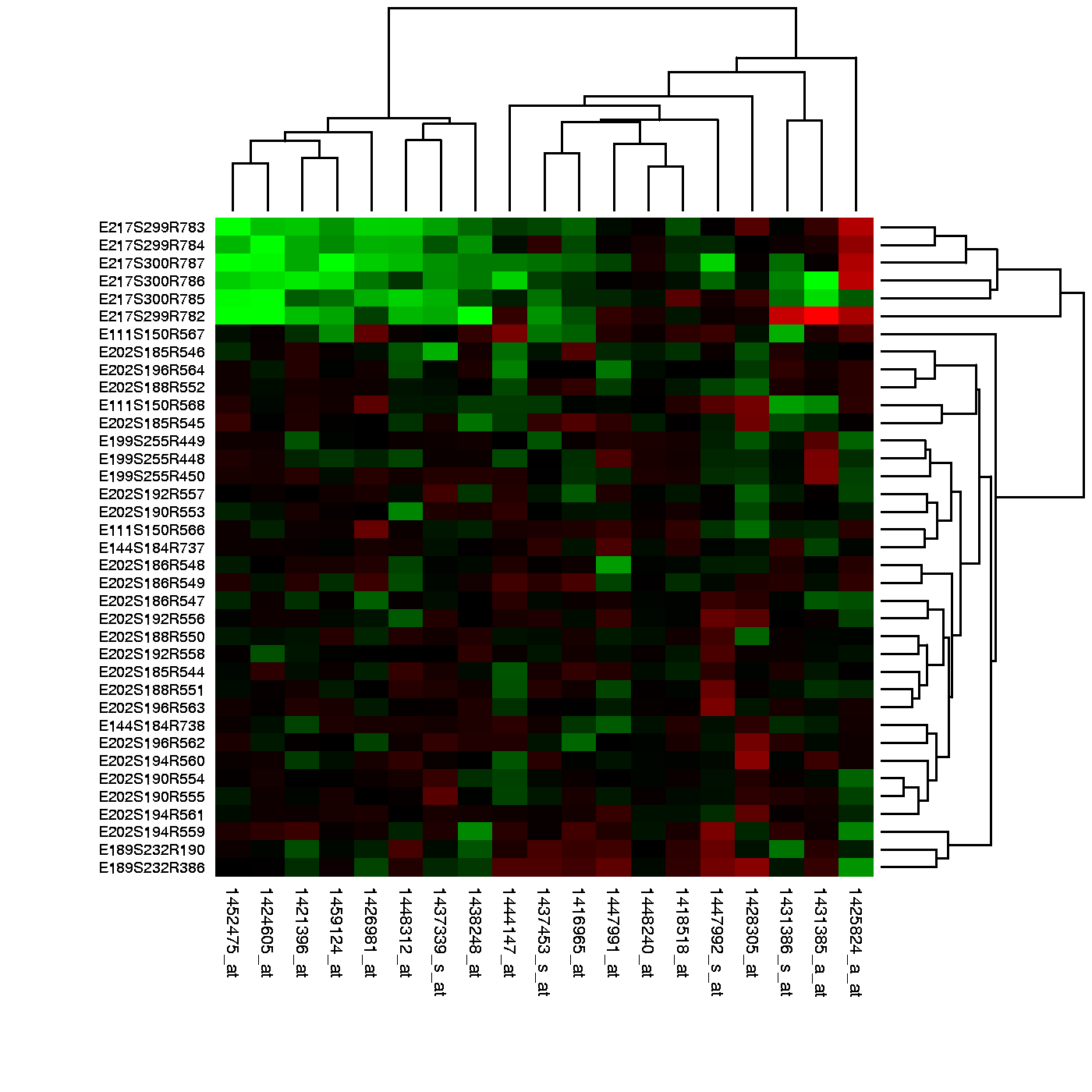
 Heat maps of gene expression values show how experimental conditions influenced production (expression) of mRNA for a set of genes. Green indicates reduced expression. Cluster analysis has placed a group of down regulated genes in the upper left corner.
Heat maps of gene expression values show how experimental conditions influenced production (expression) of mRNA for a set of genes. Green indicates reduced expression. Cluster analysis has placed a group of down regulated genes in the upper left corner.
In the field of molecular biology, gene expression profiling is the measurement of the activity (the expression) of thousands of genes at once, to create a global picture of cellular function. These profiles can, for example, distinguish between cells that are actively dividing, or show how the cells react to a particular treatment. Many experiments of this sort measure an entire genome simultaneously, that is, every gene present in a particular cell.
DNA Microarray technology[1] measures the relative activity of previously identified target genes. Sequence based techniques, like serial analysis of gene expression (SAGE, SuperSAGE) are also used for gene expression profiling. SuperSAGE is especially accurate and can measure any active gene, not just a predefined set. The advent of next-generation sequencing has made sequence based expression analysis an increasingly popular, "digital" alternative to microarrays. However, microarrays are far more common, accounting for 17,000 PubMed articles by 2006.[2]
Contents
Background
Expression profiling is a logical next step after sequencing a genome: the sequence tells us what the cell could possibly do, while the expression profile tells us what it is actually doing now. Genes contain the instructions for making messenger RNA (mRNA), but at any moment each cell makes mRNA from only a fraction of the genes it carries. If a gene is used to produce mRNA, it is considered "on", otherwise "off". Many factors determine whether a gene is on or off, such as the time of day, whether or not the cell is actively dividing, its local environment, and chemical signals from other cells. Skin cells, liver cells and nerve cells turn on (express) somewhat different genes and that is in large part what makes them different. Therefore, an expression profile allows one to deduce a cell's type, state, environment, and so forth.
Expression profiling experiments often involve measuring the relative amount of mRNA expressed in two or more experimental conditions. This is because altered levels of a specific sequence of mRNA suggest a changed need for the protein coded for by the mRNA, perhaps indicating a homeostatic response or a pathological condition. For example, higher levels of mRNA coding for alcohol dehydrogenase suggest that the cells or tissues under study are responding to increased levels of ethanol in their environment. Similarly, if breast cancer cells express higher levels of mRNA associated with a particular transmembrane receptor than normal cells do, it might be that this receptor plays a role in breast cancer. A drug that interferes with this receptor may prevent or treat breast cancer. In developing a drug, one may perform gene expression profiling experiments to help assess the drug's toxicity, perhaps by looking for changing levels in the expression of cytochrome P450 genes, which may be a biomarker of drug metabolism.[3] Gene expression profiling may become an important diagnostic test.[4][5]
Comparison to proteomics
The human genome contains on the order of 25,000 genes which work in concert to produce on the order of 1,000,000 distinct proteins. This is due to alternative splicing, and also because cells make important changes to proteins through posttranslational modification after they first construct them, so a given gene serves as the basis for many possible versions of a particular protein. In any case, a single mass spectrometry experiment can identify about 2,000 proteins[6] or 0.2% of the total. While knowledge of the precise proteins a cell makes (proteomics) is more relevant than knowing how much messenger RNA is made from each gene, gene expression profiling provides the most global picture possible in a single experiment.
Use in hypothesis generation and testing
Sometimes, a scientist already has an idea what is going on, a hypothesis, and he or she performs an expression profiling experiment with the idea of potentially disproving this hypothesis. In other words, the scientist is making a specific prediction about levels of expression that could turn out to be false.
More commonly, expression profiling takes place before enough is known about how genes interact with experimental conditions for a testable hypothesis to exist. With no hypothesis, there is nothing to disprove, but expression profiling can help to identify a candidate hypothesis for future experiments. Most early expression profiling experiments, and many current ones, have this form[7] which is known as class discovery. A popular approach to class discovery involves grouping similar genes or samples together using k-means or hierarchical clustering. The figure above represents the output of a two dimensional cluster, in which similar samples (rows, above) and similar gene probes (columns) were organized so that they would lie close together. The simplest form of class discovery would be to list all the genes that changed by more than a certain amount between two experimental conditions.
Class prediction is more difficult than class discovery, but it allows one to answer questions of direct clinical significance such as, given this profile, what is the probability that this patient will respond to this drug? This requires many examples of profiles that responded and did not respond, as well as cross-validation techniques to discriminate between them.
Limitations
In general, expression profiling studies report those genes that showed statistically significant differences under changed experimental conditions. This is typically a small fraction of the genome for several reasons. First, different cells and tissues express a subset of genes as a direct consequence of cellular differentiation so many genes are turned off. Second, many of the genes code for proteins that are required for survival in very specific amounts so many genes do not change. Third, cells use many other mechanisms to regulate proteins in addition to altering the amount of mRNA, so these genes may stay consistently expressed even when protein concentrations are rising and falling. Fourth, financial constraints limit expression profiling experiments to a small number of observations of the same gene under identical conditions, reducing the statistical power of the experiment, making it impossible for the experiment to identify important but subtle changes. Finally, it takes a great amount of effort to discuss the biological significance of each regulated gene, so scientists often limit their discussion to a subset. Newer microarray analysis techniques automate certain aspects of attaching biological significance to expression profiling results, but this remains a very difficult problem.
The relatively short length of gene lists published from expression profiling experiments limits the extent to which experiments performed in different laboratories appear to agree. Placing expression profiling results in a publicly accessible microarray database makes it possible for researchers to assess expression patterns beyond the scope of published results, perhaps identifying similarity with their own work.
Validation of high throughput measurements
Both DNA microarrays and qPCR exploit the preferential binding or "base pairing" of complementary nucleic acid sequences, and both are used in gene expression profiling, often in a serial fashion. While high throughput DNA microarrays lack the quantitative accuracy of qPCR, it takes about the same time to measure the gene expression of a few dozen genes via qPCR as it would to measure an entire genome using DNA microarrays. So it often makes sense to perform semi-quantitative DNA microarray analysis experiments to identify candidate genes, then perform qPCR on some of the most interesting candidate genes to validate the microarray results. Other experiments, such as a Western blot of some of the protein products of differentially expressed genes, make conclusions based on the expression profile more persuasive, since the mRNA levels do not necessarily correlate to the amount of expressed protein.
Statistical analysis
Data analysis of microarrays has become an area of intense research.[8] Simply stating that a group of genes were regulated by at least twofold, once a common practice, lacks a solid statistical footing. With five or fewer replicates in each group, typical for microarrays, a single outlier observation can create an apparent difference greater than two-fold. In addition, arbitrarily setting the bar at two-fold is not biologically sound, as it eliminates from consideration many genes with obvious biological significance.
Rather than identify differentially expressed genes using a fold change cutoff, one can use a variety of statistical tests or omnibus tests such as ANOVA, all of which consider both fold change and variability to create a p-value, an estimate of how often we would observe the data by chance alone. Applying p-values to microarrays is complicated by the large number of multiple comparisons (genes) involved. For example, a p-value of 0.05 is typically thought to indicate significance, since it estimates a 5% probability of observing the data by chance. But with 10,000 genes on a microarray, 500 genes would be identified as significant at p < 0.05 even if there were no difference between the experimental groups. One obvious solution is to consider significant only those genes meeting a much more stringent p value criterion, e.g., one could perform a Bonferroni correction on the p-values, or use a false discovery rate calculation to adjust p-values in proportion to the number of parallel tests involved. Unfortunately, these approaches may reduce the number of significant genes to zero, even when genes are in fact differentially expressed. Current statistics such as Rank products aim to strike a balance between false discovery of genes due to chance variation and non-discovery of differentially expressed genes. Commonly cited methods include the Significance Analysis of Microarrays (SAM)[9] and a wide variety of methods are available from Bioconductor and a variety of analysis packages from bioinformatics companies.
Selecting a different test usually identifies a different list of significant genes[10] since each test operates under a specific set of assumptions, and places a different emphasis on certain features in the data. Many tests begin with the assumption of a normal distribution in the data, because that seems like a sensible starting point and often produces results that appear more significant. Some tests consider the joint distribution of all gene observations to estimate general variability in measurements,[11] while others look at each gene in isolation. Many modern microarray analysis techniques involve bootstrapping (statistics), machine learning or Monte Carlo methods.[12]
As the number of replicate measurements in a microarray experiment increases, various statistical approaches yield increasingly similar results, but lack of concordance between different statistical methods makes array results appear less trustworthy. The MAQC Project[13] makes recommendations to guide researchers in selecting more standard methods (e.g. using p-value and fold-change together for selecting the differentially expressed genes) so that experiments performed in different laboratories will agree better.
Different from the analysis on differentially expressed individual genes, another type of analysis focuses on differential expression or perturbation of pre-defined gene sets and is called gene set analysis.[14][15] Gene set analysis demonstrated several major advantages over individual gene differential expression analysis.[14][15] Gene sets are groups of genes that are functionally related according to current knowledge. Therefore, gene set analysis is considered a knowledge based analysis approach.[14] Commonly used gene sets include those derived from KEGG pathways, Gene Ontology terms, gene groups that share some other functional annotations, such as common transcriptional regulators etc. Representative gene set analysis methods include GSEA,[14] which estimates significance of gene sets based on permutation of sample labels, and GAGE,[15] which tests the significance of gene sets based on permutation of gene labels or a parametric distribution.
Gene annotation
While the statistics may reliably identify which gene products change under experimental conditions, making biological sense of expression profiling rests on knowing which protein each gene product makes and what function this protein performs. Gene annotation provides functional and other information, for example the location of each gene within a particular chromosome. Some functional annotations are more reliable than others; some are absent. Gene annotation databases change regularly, and various databases refer to the same protein by different names, reflecting a changing understanding of protein function. Use of standardized gene nomenclature helps address the naming aspect of the problem, but exact matching of transcripts to genes[16][17] remains an important consideration.
Categorizing regulated genes
Having identified some set of regulated genes, the next step in expression profiling involves looking for patterns within the regulated set. Do the proteins made from these genes perform similar functions? Are they chemically similar? Do they reside in similar parts of the cell? Gene ontology analysis provides a standard way to define these relationships. Gene ontologies start with very broad categories, e.g., "metabolic process" and break them down into smaller categories, e.g., "carbohydrate metabolic process" and finally into quite restrictive categories like "inositol and derivative phosphorylation".
Genes have other attributes beside biological function, chemical properties and cellular location. One can compose sets of genes based on proximity to other genes, association with a disease, and relationships with drugs or toxins. The Molecular Signatures Database[18] and the Comparative Toxicogenomics Database[19] are examples of resources to categorize genes in numerous ways.
Finding patterns among regulated genes
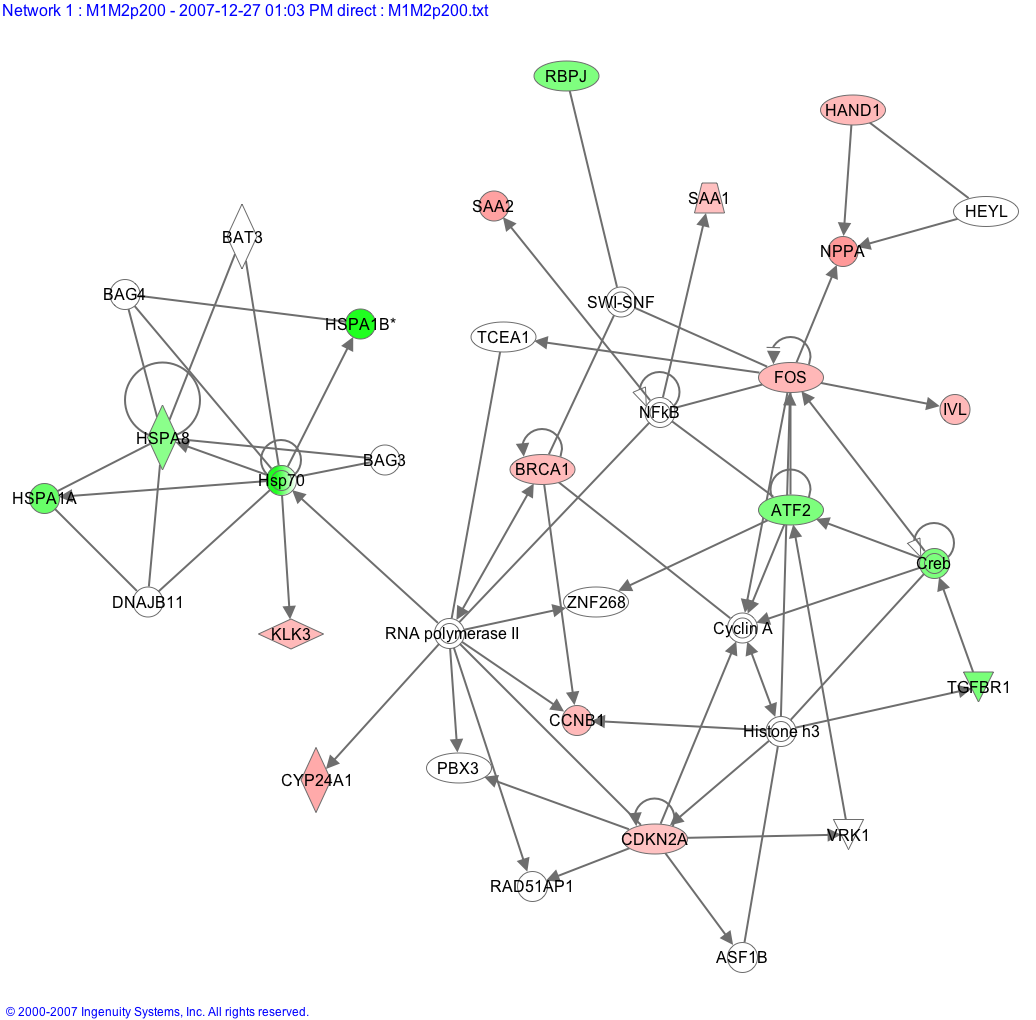
 Ingenuity Gene Network Diagram[20] which dynamically assembles genes with known relationships. Green indicates reduced expression, red indicates increased expression. The algorithm has included unregulated genes, white, to improve connectivity.
Ingenuity Gene Network Diagram[20] which dynamically assembles genes with known relationships. Green indicates reduced expression, red indicates increased expression. The algorithm has included unregulated genes, white, to improve connectivity.Regulated genes are categorized in terms of what they are and what they do, important relationships between genes may emerge.[21] For example, we might see evidence that a certain gene creates a protein to make an enzyme that activates a protein to turn on a second gene on our list. This second gene may be a transcription factor that regulates yet another gene from our list. Observing these links we may begin to suspect that they represent much more than chance associations in the results, and that they are all on our list because of an underlying biological process. On the other hand, it could be that if one selected genes at random, one might find many that seem to have something in common. In this sense, we need rigorous statistical procedures to test whether the emerging biological themes is significant or not. That is where gene set analysis[14][15] comes in.
Cause and effect relationships
Fairly straightforward statistics provide estimates of whether associations between genes on lists are greater than what one would expect by chance. These statistics are interesting, even if they represent a substantial oversimplification of what is really going on. Here is an example. Suppose there are 10,000 genes in an experiment, only 50 (0.5%) of which play a known role in making cholesterol. The experiment identifies 200 regulated genes. Of those, 40 (20%) turn out to be on a list of cholesterol genes as well. Based on the overall prevalence of the cholesterol genes (0.5%) one expects an average of 1 cholesterol gene for every 200 regulated genes, that is, 0.005 times 200. This expectation is an average, so one expects to see more than one some of the time. The question becomes how often we would see 40 instead of 1 due to pure chance.
According to the hypergeometric distribution, one would expect to try about 10^57 times (10 followed by 56 zeroes) before picking 39 or more of the chlolesterol genes from a pool of 10,000 by drawing 200 genes at random. Whether one pays much attention to how infinitesimally small the probability of observing this by chance is, one would conclude that the regulated gene list is enriched[22] in genes with a known cholesterol association.
One might further hypothesize that the experimental treatment regulates cholesterol, because the treatment seems to selectively regulate genes associated with cholesterol. While this may be true, there are a number of reasons why making this a firm conclusion based on enrichment alone represents an unwarranted leap of faith. One previously mentioned issue has to do with the observation that gene regulation may have no direct impact on protein regulation: even if the proteins coded for by these genes do nothing other than make cholesterol, showing that their mRNA is altered does not directly tell us what is happening at the protein level. It is quite possible that the amount of these cholesterol-related proteins remains constant under the experimental conditions. Second, even if protein levels do change, perhaps there is always enough of them around to make cholesterol as fast as it can be possibly made, that is, another protein, not on our list, is the rate determining step in the process of making cholesterol. Finally, proteins typically play many roles, so these genes may be regulated not because of their shared association with making cholesterol but because of a shared role in a completely independent process.
Bearing the forgoing caveats in mind, while gene profiles do not in themselves prove causal relationships between treatments and biological effects, they do offer unique biological insights that would often be very difficult to arrive at in other ways.
Using patterns to find regulated genes
As described above, one can identify significantly regulated genes first and then find patterns by comparing the list of significant genes to sets of genes known to share certain associations. One can also work the problem in reverse order. Here is a very simple example. Suppose there are 40 genes associated with a known process, for example, a predisposition to diabetes. Looking at two groups of expression profiles, one for mice fed a high carbohydrate diet and one for mice fed a low carbohydrate diet, one observes that all 40 diabetes genes are expressed at a higher level in the high carbohydrate group than the low carbohydrate group. Regardless of whether any of these genes would have made it to a list of significantly altered genes, observing all 40 up, and none down appears unlikely to be the result of pure chance: flipping 40 heads in a row is predicted to occur about one time in a trillion attempts using a fair coin.
For a type of cell, the group of genes whose combined expression pattern is uniquely characteristic to a given condition constitutes the gene signature of this condition. Ideally, the gene signature can be used to select a group of patients at a specific state of a disease with accuracy that facilitates selection of treatments.[23][24] Gene Set Enrichment Analysis (GSEA)[14] and similar methods[15] take advantage of this kind of logic but uses more sophisticated statistics, because component genes in real processes display more complex behavior than simply moving up or down as a group, and the amount the genes move up and down is meaningful, not just the direction. In any case, these statistics measure how different the behavior of some small set of genes is compared to genes not in that small set.
GSEA uses a Kolmogorov Smirnov style statistic to see whether any previously defined gene sets exhibited unusual behavior in the current expression profile. This leads to a multiple hypothesis testing challenge, but reasonable methods exist to address it.[25]
Conclusions
Expression profiling provides new information about what genes do under various conditions. Overall, microarray technology produces reliable expression profiles.[26] From this information one can generate new hypotheses about biology or test existing ones. However, the size and complexity of these experiments often results in a wide variety of possible interpretations. In many cases, analyzing expression profiling results takes far more effort than performing the initial experiments.
Most researchers use multiple statistical methods and exploratory data analysis before publishing their expression profiling results, coordinating their efforts with a bioinformatician or other expert in microarray technology. Good experimental design, adequate biological replication and follow up experiments play key roles in successful expression profiling experiments.
See also
- Gene expression profiling in cancer
References
- ^ "Microarrays Factsheet". http://www.ncbi.nlm.nih.gov/About/primer/microarrays.html. Retrieved 2007-12-28.
- ^ Kawasaki ES (July 2006). "The end of the microarray Tower of Babel: will universal standards lead the way?" (– Scholar search). J Biomol Tech 17 (3): 200–6. PMC 2291790. PMID 16870711. http://jbt.abrf.org/cgi/pmidlookup?view=long&pmid=16870711.[dead link]
- ^ Suter L, Babiss LE, Wheeldon EB (2004). "Toxicogenomics in predictive toxicology in drug development". Chem. Biol. 11 (2): 161–71. doi:10.1016/j.chembiol.2004.02.003. PMID 15123278.
- ^ Magic Z, Radulovic S, Brankovic-Magic M (2007). "cDNA microarrays: identification of gene signatures and their application in clinical practice". J BUON 12 Suppl 1: S39–44. PMID 17935276.
- ^ Cheung AN (2007). "Molecular targets in gynaecological cancers". Pathology 39 (1): 26–45. doi:10.1080/00313020601153273. PMID 17365821.
- ^ Mirza SP, Olivier M (2007). "Methods and approaches for the comprehensive characterization and quantification of cellular proteomes using mass spectrometry". Physiol Genomics 33 (1): 3. doi:10.1152/physiolgenomics.00292.2007. PMC 2771641. PMID 18162499. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2771641.
- ^ Chen JJ (2007). "Key aspects of analyzing microarray gene-expression data". Pharmacogenomics 8 (5): 473–82. doi:10.2217/14622416.8.5.473. PMID 17465711.
- ^ Vardhanabhuti S, Blakemore SJ, Clark SM, Ghosh S, Stephens RJ, Rajagopalan D (2006). "A comparison of statistical tests for detecting differential expression using Affymetrix oligonucleotide microarrays". OMICS 10 (4): 555–66. doi:10.1089/omi.2006.10.555. PMID 17233564.
- ^ "Significance Analysis of Microarrays". http://www-stat.stanford.edu/~tibs/SAM/. Retrieved 2007-12-27.
- ^ Yauk CL, Berndt ML (2007). "Review of the literature examining the correlation among DNA microarray technologies". Environ. Mol. Mutagen. 48 (5): 380–94. doi:10.1002/em.20290. PMC 2682332. PMID 17370338. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2682332.
- ^ Breitling R (2006). "Biological microarray interpretation: the rules of engagement". Biochim. Biophys. Acta 1759 (7): 319–27. doi:10.1016/j.bbaexp.2006.06.003. PMID 16904203.
- ^ Draminski M, Rada-Iglesias A, Enroth S, Wadelius C, Koronacki J, Komorowski J (2008). "Monte Carlo feature selection for supervised classification". Bioinformatics 24 (1): 110–7. doi:10.1093/bioinformatics/btm486. PMID 18048398.
- ^ Dr. Leming Shi, National Center for Toxicological Research. "MicroArray Quality Control (MAQC) Project". U.S. Food and Drug Administration. http://www.fda.gov/nctr/science/centers/toxicoinformatics/maqc/. Retrieved 2007-12-26.
- ^ a b c d e f Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005). "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles". Proc. Natl. Acad. Sci. U.S.A. 102 (43): 15545–50. doi:10.1073/pnas.0506580102. PMC 1239896. PMID 16199517. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1239896.
- ^ a b c d e Luo W, Friedman M, Shedden K, Hankenson KD, Woolf JP (2009). "GAGE: generally applicable gene set enrichment for pathway analysis". BMC Bioinformatics 10: 161. doi:10.1186/1471-2105-10-161. PMC 2696452. PMID 19473525. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2696452.
- ^ Dai M, Wang P, Boyd AD, et al. (2005). "Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data". Nucleic Acids Res. 33 (20): e175. doi:10.1093/nar/gni179. PMC 1283542. PMID 16284200. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1283542.
- ^ Alberts R, Terpstra P, Hardonk M, et al. (2007). "A verification protocol for the probe sequences of Affymetrix genome arrays reveals high probe accuracy for studies in mouse, human and rat". BMC Bioinformatics 8: 132. doi:10.1186/1471-2105-8-132. PMC 1865557. PMID 17448222. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1865557.
- ^ "GSEA – MSigDB". http://www.broad.mit.edu/gsea/msigdb/index.jsp. Retrieved 2008-01-03.
- ^ "CTD: The Comparative Toxicogenomics Database". http://ctdbase.org/. Retrieved 2008-01-03.
- ^ "Ingenuity Systems". http://www.ingenuity.com. Retrieved 2007-12-27.
- ^ Alekseev OM, Richardson RT, Alekseev O, O'Rand MG (2009). "Analysis of gene expression profiles in HeLa cells in response to overexpression or siRNA-mediated depletion of NASP". Reprod. Biol. Endocrinol. 7: 45. doi:10.1186/1477-7827-7-45. PMC 2686705. PMID 19439102. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2686705.
- ^ Curtis RK, Oresic M, Vidal-Puig A (2005). "Pathways to the analysis of microarray data". Trends Biotechnol. 23 (8): 429–35. doi:10.1016/j.tibtech.2005.05.011. PMID 15950303.
- ^ Mook S, Van't Veer LJ, Rutgers EJ, Piccart-Gebhart MJ, Cardoso F (2007). "Individualization of therapy using Mammaprint: from development to the MINDACT Trial". Cancer Genomics Proteomics 4 (3): 147–55. PMID 17878518.
- ^ Corsello SM, Roti G, Ross KN, Chow KT, Galinsky I, DeAngelo DJ, Stone RM, Kung AL, Golub TR, Stegmaier K (June 2009). "Identification of AML1-ETO modulators by chemical genomics". Blood 113 (24): 6193–205. doi:10.1182/blood-2008-07-166090. PMC 2699238. PMID 19377049. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2699238.
- ^ "GSEA". http://www.broad.mit.edu/gsea/msigdb/index.jsp. Retrieved 2008-01-09.
- ^ Couzin J (2006). "Genomics. Microarray data reproduced, but some concerns remain". Science 313 (5793): 1559. doi:10.1126/science.313.5793.1559a. PMID 16973852.
Categories:- Molecular genetics
- Microarrays
Wikimedia Foundation. 2010.