- Molecular anthropology
-
Molecular anthropology is a field of anthropology in which molecular analysis is used to determine evolutionary links between ancient and modern human populations, as well as between contemporary species. Generally, comparisons are made between sequence, either DNA or protein sequence, however early studies used comparative serology.
By examining DNA sequences in different populations, scientists can determine the closeness of relationships between populations (or within populations). Certain similarities in genetic makeup let molecular anthropologists determine whether or not different groups of people belong to the same haplogroup, and thus if they share a common geographical origin. This is significant because it allows anthropologists to trace patterns of migration and settlement, which gives helpful insight as to how contemporary populations have formed and progressed over time.[1]
Molecular anthropology has been extremely useful in establishing the evolutionary tree of humans and other primates, including closely related species like chimps and gorillas. While there are clearly many morphological similarities between humans and chimpanzees, for example, certain studies also have concluded that there is roughly a 98 percent commonality between the DNA of both species.[citation needed] However, more recent studies have modified the commonality of 98 percent to a commonality of 94 percent, showing that the genetic gap between humans and chimps is bigger than originally thought.[2] Such information is useful in searching for common ancestors and coming to a better understanding of how humans evolved.
Contents
Haploid loci in molecular anthropology
 Image of mitochondrion. There are many mitochondria within a cell, and DNA in them replicates independently of the chromosomes in the nucleus.
Image of mitochondrion. There are many mitochondria within a cell, and DNA in them replicates independently of the chromosomes in the nucleus.
There are two continuous linkage groups in human that are carried by a single sex. The first is the Y chromosome, which is passed from father to son. Anatomical females carry a Y chromosome only rarely, as a result of genetic defect. The other linkage group is the mitochondrial DNA (mtDNA). MtDNA can only be passed to the next generation by females but only under highly exceptional circumstances is mtDNA passed through males.[clarification needed] The non-recombinant portion of the Y chromosome and the mtDNA, under normal circumstances, do not undergo productive recombination. Part of the Y chromosome can undergo recombination with the X chromosome and within ape history the boundary has changed. Such recombinant changes in the non-recombinant region of Y are extremely rare.[citation needed]
Mitochondrial DNA
 Illustration showing mitochondrial DNA; the control region (D-loop, hypervariable regions 1 and II are located on left and right side at the top, respectively).
Illustration showing mitochondrial DNA; the control region (D-loop, hypervariable regions 1 and II are located on left and right side at the top, respectively).Mitochondrial DNA became an area of research in phylogenetics in the late 1970s. Unlike genomic DNA, it offered advantages in that it did not undergo recombination. The process of recombination, if frequent enough, corrupts the ability to create parsimonious trees because stretches of amino acid subsititions (SNPs).[clarification needed] When looking between distantly related species, recombination is less of a problem since recombination between branches from common ancestors is prevented after true speciation occurs. When examining closely related species, or branching within species, recombination creates a large number of 'irrelevant SNPs' for cladistic analysis. MtDNA, through the process of organelle division, become clonal over time; very little, too often none, of that paternal mtDNA is passed. While recombination may occur in mtDNA, there is little risk that it will be passed to the next generation. As a result, mtDNA become clonal copies of each other, except when a new mutation arises. As a result, mtDNA does not have pitfalls of autosomal loci when studied in interbreeding groups. Another advantage of mtDNA is that the hyper-variable regions evolve very quickly; this exhibits that certain regions of mitochondrial DNA approach neutrality. This allowed the use of mitochondrial DNA to determine that the relative age of the human population was small, having gone through a recent constriction at about 150,000 years ago (see Sources of error).
Mitochondrial DNA has also been used to verify the proximity of chimpanzees to humans relative to gorillas, and to verify the relationship of these three species relative to orangutan.
 A population bottleneck, as illustrated was detected by intrahuman mtDNA phylogenetic studies; the length of the bottleneck itself is indeterminate per mtDNA.
A population bottleneck, as illustrated was detected by intrahuman mtDNA phylogenetic studies; the length of the bottleneck itself is indeterminate per mtDNA.More recently,[when?] the mtDNA genome has been used to estimate branching patterns in peoples around the world, such as when the new world was settled and how. The problem with these studies have been that they rely heavily on mutations in the coding region. Researchers have increasingly discovered that as humans moved from Africa's south-eastern regions, that more mutations accumulated in the coding region than expected, and in passage to the new world some groups are believed[citation needed] to have passed from the Asian tropics to Siberia to an ancient land region called Beringia and quickly migrated to South America. Many of the mtDNA have far more mutations and at rarely mutated coding sites relative to expectations of neutral mutations.
Mitochondrial DNA offers another advantage over autosomal DNA. There are generally 2 to 4 copies of each chromosome in each cell (1 to 2 from each parent chromosome). For mtDNA there can be dozens to hundreds in each cell. This increases the amount of each mtDNA loci by at least a magnitude. For ancient DNA, in which the DNA is highly degraded, the number of copies of DNA is helpful in extending and bridging short fragments together, and decreases the amount of bone extracted from highly valuable fossil/ancient remains. Unlike Y chromosome, both male and female remains carry mtDNA in roughly equal quantities.
 Schematic of typical animal cell, showing subcellular components. Organelles: (1) nucleolus (2) nucleus (9) mitochondria
Schematic of typical animal cell, showing subcellular components. Organelles: (1) nucleolus (2) nucleus (9) mitochondriaY chromosome
 Illustration of human Y chromosome
Illustration of human Y chromosomeThe Y chromosome is found in the nucleus of normal cells (nuclear DNA). Unlike mtDNA, it has mutations in the non-recombinant portion (NRY) of the chromosome spaced widely apart, so far apart that finding the mutations on new Y chromosomes is labor intensive compared with mtDNA. Many studies rely on tandem repeats; however, tandem repeats can expand and retract rapidly and in some predictable patterns. The Y chromosome only tracks male lines, and is not found in females, whereas mtDNA can be traced in males even though they fail to pass on mtDNA. In addition, it has been estimated that effective male populations in the prehistoric period were typically two females per male, and recent studies show that cultural hegemony plays a large role in the passage of Y. This has created discordance between males and females for the Time to the Most Recent Common Ancestor (TMRCA). The estimates for Y TMRCA range from 1/4 to less than 1/2 that of mtDNA TMRCA. It is unclear whether this is due to high male-to-female ratios in the past coupled with repeat migrations from Africa, as a result of mutational rate change, or as some have even proposed that females of the LCA between chimps and humans continued to pass DNA millions after males ceased to pass DNA. At present the best evidence suggests that in migration the male to female ratio in humans may have declined, causing a trimming of Y diversity on multiple occasions within and outside of Africa.
 Diagram of human X chromosome showing genetic map
Diagram of human X chromosome showing genetic mapFor short-range molecular phylogenetics and molecular clocking, the Y chromosome is highly effective and creates a second perspective. One argument that arose was that the Maori by mtDNA appear to have migrated from Eastern China or Taiwan, by Y chromosome from the Papua New Guinea region. When HLA haplotypes were used to evaluate the two hypotheses, it was uncovered that both were right, that the Maori were an admixed population. Such admixtures appear to be common in the human population and thus the use of a single haploid loci can give a biased perspective.
X-linked Studies
The X-chromosome is also a form of nuclear DNA. Since it is found as 1 copy in males and 2 non-identical chromosomes in females it has a ploidy of 1.5. However, in humans the effective ploidy is somewhat higher, ~1.7, as females in the breeding population have tended to outnumber males by 2:1 during a large portion of human prehistory. Like mtDNA, X-linked DNA tends to over emphasize female population history much more than male. There have been several studies of loci on X chromosome, in total 20 sites have been examined. These include PDHA1, PDHA1, Xq21.3, Xq13.3, Zfx, Fix, Il2rg, Plp, Gk, Ids, Alas2, Rrm2p4, AmeIX, Tnfsf5, Licam, and Msn. The time to most recent common ancestor (TMRCA) ranges from fixed to ~1.8 million years, with a median around 700ky. These studies roughly plot to the expected fixation distribution of alleles, given linkage disequilibrium between adjacent sites. For some alleles the point of origin is elusive, for others, the point of origin points toward Sub-Saharan Africa. There are some distinctions within SSA that suggest a smaller region, but there is not adequate enough sample size and coverage to define a place of most recent common ancestor. The TMRCA is consistent with and extends the bottleneck implied by mtDNA, confidently to about 500,000 years.
Autosomal Loci
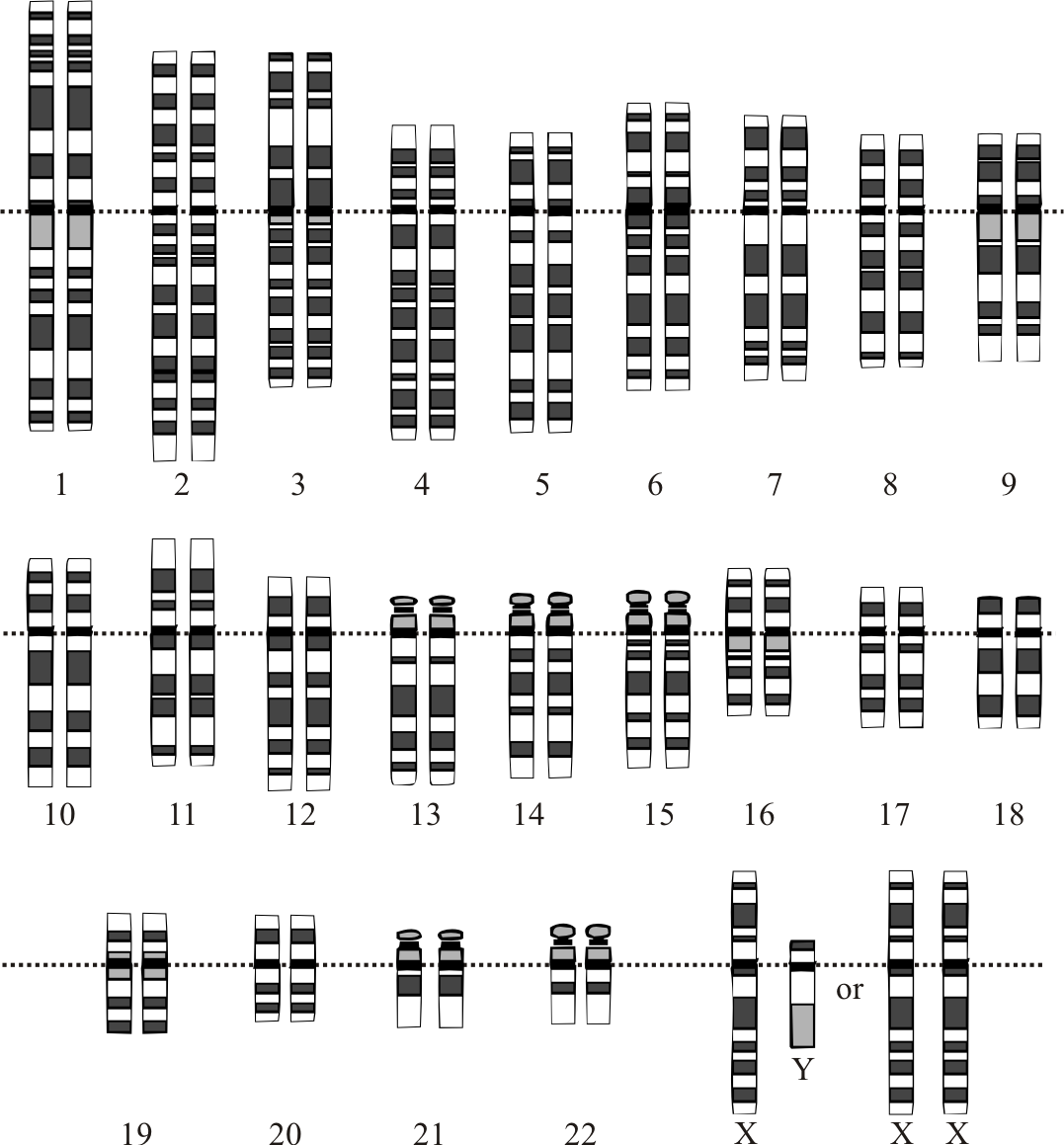
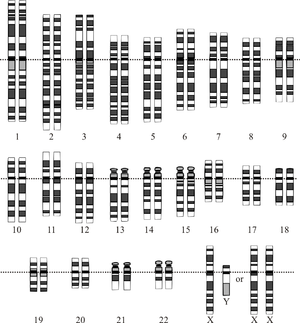 Diagram of human karyotype
Diagram of human karyotypeRate variation
Ancient DNA sequencing
Since Krings Neandertal mtDNA have been sequenced, and the sequence similarity indicates an equally recent origin from a small population on the Neanderthal branch of late hominids. MCR1 gene has also been sequenced but the results are controversial, with one study claiming that contamination issues cannot be resolved from human Neandertal similarities. Critically however no DNA sequence has been obtained from Homo erectus, Homo floriensis, or any of the other late hominids. Some of the ancient sequences obtained have highly probable errors, and proper control to avoid contamination.
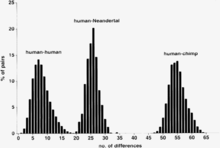 Comparison of differences between human and Neanderthal mtDNA
Comparison of differences between human and Neanderthal mtDNACauses of errors
The molecular phylogenetics is based on quantification substitutions and then comparing sequence with other species, there are several points in the process which create errors. The first and greatest challenge is finding "anchors" that allow the research to calibrate the system. In this example, there are 10 mutations between chimp and humans, but the researcher has no known fossils that are agreeably ancestral to both but not ancestral to the next species in the tree, gorilla. However, there are fossils believed to be ancestral to Orangutans and Humans, from about 14 million years ago. So that the researcher can use Orangutan and Human comparison and comes up with a difference of 24. Using this he can estimate (24/(14*2, the "2" is for the length of the branch to Human (14my) and the branch to Orangutan (14 my) from their last common ancestor (LCA). The mutation rate at 0.857 for a stretch of sequence. Mutation rates are given, however, as rate per nucleotide(nt)-site, so if the sequence were say 100 nt in length that rate would be 0.00857/nt per million years. Ten mutations*100nt/(0.00857*2) = 5.8 million years.
Problem of calibration
There are several problems not seen in the above. First, mutations occur as random events. Second, the chance that any site in the genome varies is different from the next site, a very good example is the codons for amino acids, the first two nt in a codon may mutate at 1 per billion years, but the third nt may mutate 1 per million years. Unless scientist study the sequence of a great many animals, particularly those close to the branch being examined, they generally do not know what the rate of mutation for a given site. Mutations do occur at 1st and 2nd positions of codons, but in most cases these mutations are under negative selection and so are removed from the population over small periods of time. In defining the rate of evolution in the anchor one has the problem that random mutation creates. For example a rate of .005 or .010 can also explain 24 mutations according to the binomial probability distribution. Some of the mutations that did occur between the two have reverted, hiding an initially higher rate. Selection may play into this, a rare mutation may be selective at point X in time, but later climate may change or the species migrates and it is not longer selective, and pressure exerted on new mutations that revert the change, and sometimes the reversion of a nt can occur, the greater the distance between two species the more likely this is going to occur. In addition, from that ancestral species both species may randomly mutate a site to the same nucleotide. Many times this can be resolved by obtaining DNA samples from species in the branches, creating a parsimonious tree in which the order of mutation can be deduced, creating branch-length diagram. This diagram will then produce a more accurate estimate of mutations between two species. Statistically one can assign variance based on the problem of randomnicity, back mutations, and parallel mutations (homoplasies) in creating an error range.
There is another problem in calibration however that has defied statistical analysis. There is a true/false designation of a fossil to a least common ancestor. In reality the odds of having the least common ancestor of two extant species as an anchor is low, often that fossil already lies in one branch (underestimating the age), lies in a third branch (underestimating the age) or in the case of being within the LCA species, may have been millions of years older than the branch. To date the only way to assess this variance is to apply molecular phylogenetics on species claimed to be branch points. This only, however identifies the 'outlying' anchor points. And since it is more likely the more abundant fossils are younger than the branch point the outlying fossil may simply be a rare older representative. These unknowns create uncertainty that is difficult to quantify, and often not attempted.
Recent papers have been able to estimate, roughly, variance. The general trend as new fossils are discovered, is that the older fossils underestimated the age of the branch point. In addition to this dating of fossils has had a history of errors and there have been many revised datings. The age assigned by researchers to some major branch points have almost doubled in age over the last 30 years. An excellent example of this is the debate over LM3 (Mungo lake 3) in Australia. Originally it was dated to around 30 ky by carbon dating, carbon dating has problems, however, for sampled over 20ky in age, and severe problems for samples around 30ky in age. Another study looked at the fossil and estimated the age to be 62 ky in age.
At the point one has an estimation of mutation rate, given the above there must be two sources of variance that need to be cross-multiplied to generate an overall variance. This is infrequently done in the literature.
Problems in estimating TMRCA
Time to most recent common ancestor (TMRCA) combines the errors in calibration with errors in determining the age of a local branch.
History
Protein era
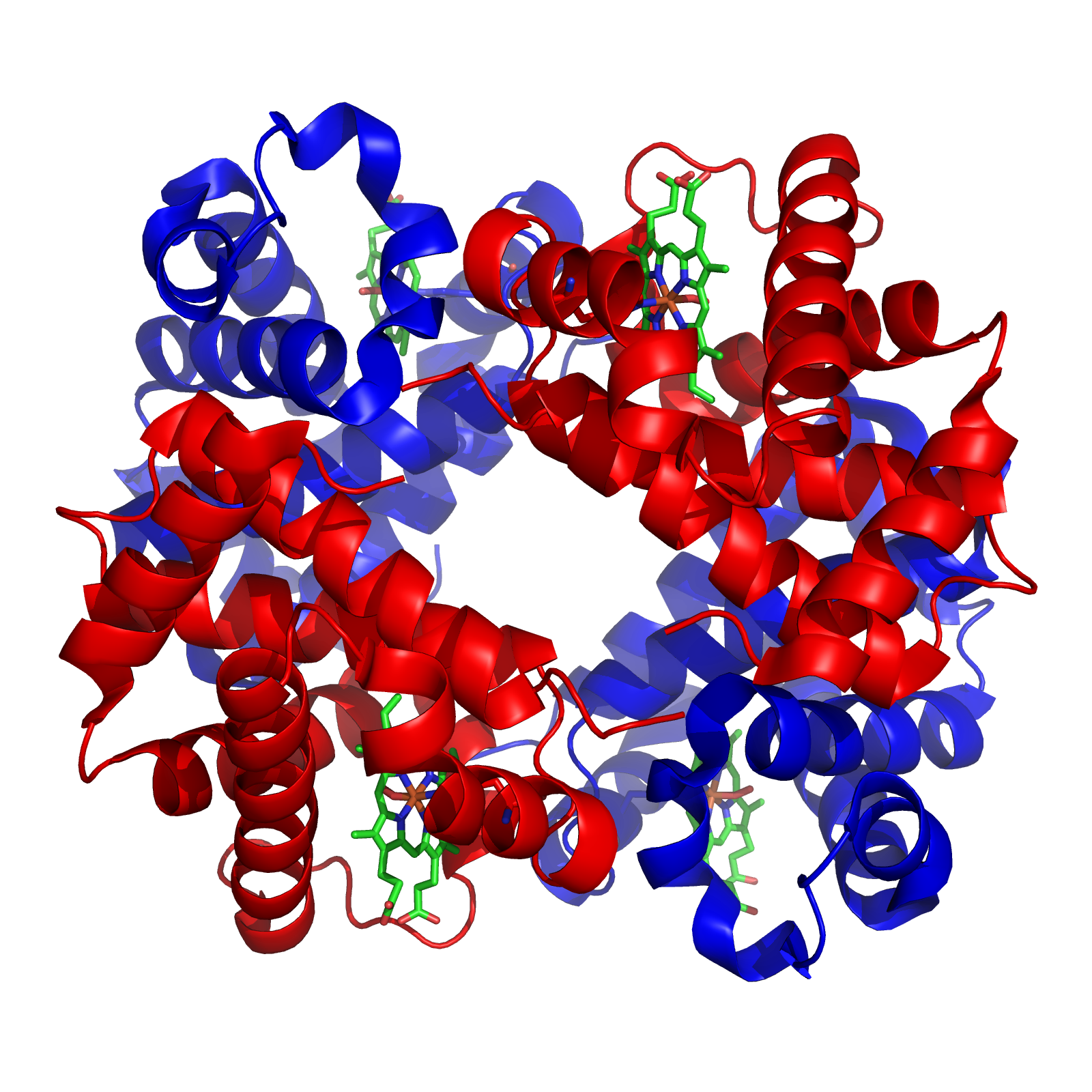
 Structure of human hemoglobin. Hemoglobins from dozens of animals and even plants were sequenced in the 1960s and early 1970s
Structure of human hemoglobin. Hemoglobins from dozens of animals and even plants were sequenced in the 1960s and early 1970sWith DNA newly discovered as the genetic material, in the early 1960s protein sequencing was beginning to take off.[3] Protein sequencing began on cytochrome C and Hemoglobin. Gerhard Braunitzer sequenced hemoglobin and myoglobin, in total more than hundreds of sequences from wide ranging species were done. In 1967 A.C. Wilson began to promote the idea of a "molecular clock". By 1969 molecular clocking was applied to anthropoid evolution and V. Sarich and A.C. Wilson found that albumin and hemoglobin has comparable rates of evolution, indicating chimps and humans split about 4 to 5 million years ago.[4] In 1970, Louis Leakey confronted this conclusion with arguing for improper calibration of molecular clocks.[5] By 1975 protein sequencing and comparative serology combined were used to propose that humans closest living relative (as a species) was the chimpanzee.[6] In hindsight, the last common ancestor (LCA) from humans and chimps appears to older than the Sarich and Wilson estimate, but not as old as Leakey claimed , either. However, Leakey was correct in the divergence of old and new world monkeys, the value Sarich and wilson used was a significant underestimate. This error in prediction capability highlights a common theme. (See Causes of Error)
DNA era
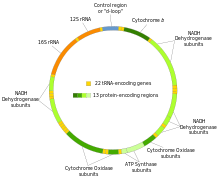 Restriction fragment length polymorphisms studies the cutting of mtDNA into fragments, Later the focus of PCR would be on the D 'contol'-loop, at the top of the circle
Restriction fragment length polymorphisms studies the cutting of mtDNA into fragments, Later the focus of PCR would be on the D 'contol'-loop, at the top of the circleRLFP and DNA hybridization
In 1979, W.M.Brown and Wilson began looking at the evolution of mitochodrial DNA in animals, and found they were evolving rapidly.[7] The technique they used was restriction fragment length polymorphism (RFLP), which was more affordable at the time compared to sequencing. In 1980, W.M. Brown, looking at the relative variation between human and other species, recognized there was a recent constriction (180,000 years ago) in the human population.[8] A year later Brown and Wilson were looking at RFLP fragments and determined the human population expanded more recently than other ape populations.[9] In 1984 the first DNA sequence from an extinct animal was done.[10] Sibley and Ahlquist apply DNA-DNA hybridization technology to anthropoid phylogeny, and see pan/human split closer than gorilla/pan or gorilla/human split, a highly controversial claim.[11][12] However, in 1987 they were able to support their claim.[13] In 1987, Cann, Stoneking and Wilson suggest, by RFLP analysis of human mitochondrial DNA, that humans evolved from a constrict in Africa of a single female in a small population, ~10,00 individuals, 200,000 years ago.[14]
Era of PCR
 PCR could rapidly amplify DNA from one molecule to billions, allowing sequencing from human hairs or ancient DNA.
PCR could rapidly amplify DNA from one molecule to billions, allowing sequencing from human hairs or ancient DNA.In 1987, PCR-amplification of mtDNA was first used to determine sequences.[15] In 1991 Vigilante et al. published the seminal work on mtDNA phylogeny implicating sub-saharan Africa as the place of humans most recent common ancestors for all mtDNAs.[16] The war between out-of-Africa and multiregionalism, already simmering with the critiques of Allan Templeton, soon escalated with the paleoanthropologist, like Milford Wolpoff, getting involved.[17][18][19] In 1995, F. Ayala published his critical Science article "The Myth about Eve", which relied on HLA-DR sequence.[20] At the time, however Ayala was not aware of rapid evolution of HLA loci via recombinatory process. In 1996, Parham and Ohta published their finds on the rapid evolution of HLA by short-distance recombination ('gene conversion' or 'abortive recombination'), weakening Ayala's claim (Parham had actually written a review a year earlier, but this had gone unnoticed).[21][22] A stream of papers would follow from both sides, many with highly flawed methods and sampling. One of the more interesting[says who?] was Harris and Hey, 1998 which showed that the TMCRA (time to most recent common ancestor) for the PDHA1 gene was well in excess of 1 million years. Given a ploidy at this locus of 1.5 (3 fold higher than mtDNA) the TMRCA was more than double the expectation. While this falls into the 'fixation curve' of 1.5 ploidy (averaging 2 female and 1 male) the suggested age of 1.8 my is close a significantly deviant p-value for the population size, possibly indicating that the human population shrank or split off of another population.[23] Oddly, the next X-linked loci they examined, Factor IX, showed a TMRCA of less than 300,000 years.[24]
 Cross-linked DNA extracted from the 4,000-year-old liver of an Ancient Egyptian priest Called Nekht-Ankh
Cross-linked DNA extracted from the 4,000-year-old liver of an Ancient Egyptian priest Called Nekht-AnkhAncient DNA
Ancient DNA sequencing had been conducted on a limited scale up to the late 1990s when the staff at the Max Planck Institute shocked the anthropology world by sequencing DNA from an estimated 40,000-year-old Neanderthal.[25][26][27] The result of that experiment is that the differences between humans living in Europe, many of which were derived from haplogroup H (CRS), Neandertals branched from humans more than 300,000 years before haplogroup H reached Europe. While the mtDNA and other studies continued to support a unique recent African origin, this new study basically answered critiques from the Neandertal side.
Genomic sequencing
Significant progress has been made in genomic sequencing since Ingman and colleague published their finding on mitochondrial genome.[28] Several papers on genomic mtDNA have been published; there is considerable variability in the rate of evolution, and rate variation and selection are evident at many sites. In 2007, Gonder et al. proposed that a core population of humans, with greatest level of diversity and lowest selection, once lived in the region of Tanzania and proximal parts of southern Africa, since humans left this part of Africa, mitochondria have been selectively evolving to new regions.[29]
Critical Progress
Critical in the history of molecular anthropology:
- That molecular phylogenetics could compete with comparative anthropology for determining the proximity of species to humans.
- Wilson and King realized in 1975, that while there was equity between the level of molecular evolution branching from chimp to human to putative LCA, that there was an inequity in morphological evolution. Comparative morphology based on fossils could be biased by different rates of change.[6]
- Realization that in DNA there are multiple independent comparisons. Two techniques, mtDNA and hybridization converge on a single answer, chimps as a species are most closely related to humans.
- The ability to resolve population sizes based on the 2N rule, proposed by Kimura in the 1950s.[30] To use that information to compare relative sizes of population and come to a conclusion about abundance that contrasted observations based on the paleontological record. While human fossils in the early and middle stone age are far more abundant than Chimpazee or Gorilla, there are few unambiguous chimpanzee or gorilla fossils from the same period
Loci that have been used in molecular phylogenetics:
- Cytochrome C
- Serum Albumin
- Hemoglobin - Braunitizer, 1960s, Harding et al. 1997
- Mitochondrial D-loop - Wilson group, 1980, 1981, 1984, 1987, 1989, 1991(posthumously) - TMRCA about 170 kya.
- Y-chromosome
- HLA-DR - Ayala 1995 - TMRCA for locus is 60 million years.
- CD4 (Intron) - Tishkoff, 1996 - most of the diversity is in Africa.
- PDHA1 (X-linked) Harris and Hey - TMRCA for locus greater than 1.5 million years.
Xlinked loci: PDHA1, Xq21.3, Xq13.3, Zfx, Fix, Il2rg, Plp, Gk, Ids, Alas2, Rrm2p4, AmeIX, Tnfsf5, Licam, and Msn
Autosomal:Numerous.References
- ^ Kottak, Conrad Phillip. Windows on Humanity. New York: McGraw-Hill, 2005.
- ^ "Humans and Chimps: Close But Not That Close". Scientific American. 2006-12-19. http://www.sciam.com/article.cfm?chanID=sa003&articleID=9D0DAC2B-E7F2-99DF-3AA795436FEF8039. Retrieved 2006-12-20.
- ^ A.C.Wilson and N.O.Kaplan (1963) Enzymes and nucleic acids in systematics. Proceedings of the XVI International Congress of Zoology Vol.4, pp.125-127.
- ^ Wilson AC, Sarich VM (August 1969). "A molecular time scale for human evolution". Proc. Natl. Acad. Sci. U.S.A. 63 (4): 1088–93. Bibcode 1969PNAS...63.1088W. doi:10.1073/pnas.63.4.1088. PMC 223432. PMID 4982244. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=223432.
- ^ Leakey LS (October 1970). "The relationship of African apes, man and old world monkeys". Proc. Natl. Acad. Sci. U.S.A. 67 (2): 746–8. Bibcode 1970PNAS...67..746L. doi:10.1073/pnas.67.2.746. PMC 283268. PMID 5002096. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=283268.
- ^ a b King MC, Wilson AC (April 1975). "Evolution at two levels in humans and chimpanzees". Science 188 (4184): 107–16. Bibcode 1975Sci...188..107K. doi:10.1126/science.1090005. PMID 1090005. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=1090005.
- ^ Brown WM, George M, Wilson AC (April 1979). "Rapid evolution of animal mitochondrial DNA". Proc. Natl. Acad. Sci. U.S.A. 76 (4): 1967–71. Bibcode 1979PNAS...76.1967B. doi:10.1073/pnas.76.4.1967. PMC 383514. PMID 109836. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=383514.
- ^ Brown WM (June 1980). "Polymorphism in mitochondrial DNA of humans as revealed by restriction endonuclease analysis". Proc. Natl. Acad. Sci. U.S.A. 77 (6): 3605–9. Bibcode 1980PNAS...77.3605B. doi:10.1073/pnas.77.6.3605. PMC 349666. PMID 6251473. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=349666.
- ^ Ferris SD, Brown WM, Davidson WS, Wilson AC (October 1981). "Extensive polymorphism in the mitochondrial DNA of apes". Proc. Natl. Acad. Sci. U.S.A. 78 (10): 6319–23. Bibcode 1981PNAS...78.6319F. doi:10.1073/pnas.78.10.6319. PMC 349030. PMID 6273863. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=349030.
- ^ Higuchi R, Bowman B, Freiberger M, Ryder OA, Wilson AC (1984). "DNA sequences from the quagga, an extinct member of the horse family". Nature 312 (5991): 282–4. Bibcode 1984Natur.312..282H. doi:10.1038/312282a0. PMID 6504142.
- ^ Sibley CG, Ahlquist JE (1984). "The phylogeny of the hominoid primates, as indicated by DNA-DNA hybridization". J. Mol. Evol. 20 (1): 2–15. doi:10.1007/BF02101980. PMID 6429338.
- ^ Templeton AR (September 1985). "The phylogeny of the hominoid primates: a statistical analysis of the DNA-DNA hybridization data". Mol. Biol. Evol. 2 (5): 420–33. PMID 3939706. http://mbe.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=3939706.
- ^ Sibley CG, Ahlquist JE (1987). "DNA hybridization evidence of hominoid phylogeny: results from an expanded data set". J. Mol. Evol. 26 (1–2): 99–121. doi:10.1007/BF02111285. PMID 3125341.
- ^ Cann RL, Stoneking M, Wilson AC (1987). "Mitochondrial DNA and human evolution". Nature 325 (6099): 31–6. Bibcode 1987Natur.325...31C. doi:10.1038/325031a0. PMID 3025745.
- ^ Wrischnik LA, Higuchi RG, Stoneking M, Erlich HA, Arnheim N, Wilson AC (January 1987). "Length mutations in human mitochondrial DNA: direct sequencing of enzymatically amplified DNA". Nucleic Acids Res. 15 (2): 529–42. doi:10.1093/nar/15.2.529. PMC 340450. PMID 2881260. http://nar.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=2881260.
- ^ Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC (September 1991). "African populations and the evolution of human mitochondrial DNA". Science 253 (5027): 1503–7. Bibcode 1991Sci...253.1503V. doi:10.1126/science.1840702. PMID 1840702. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=1840702.
- ^ Templeton AR. The 'Eve' Hypothesis: A genetic critique and reanalysis. American Anthropologist 95: 51-72.
- ^ Thorne A and Wolpoff M. The multiregional evolution of Humans. Scientific American (April) pp. 28-33 (1992)
- ^ Wolpoff M and Thorne A. The case against Eve. New Scientist (1991) pp. 37-41.
- ^ Ayala FJ (December 1995). "The myth of Eve: molecular biology and human origins". Science 270 (5244): 1930–6. Bibcode 1995Sci...270.1930A. doi:10.1126/science.270.5244.1930. PMID 8533083. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=8533083.
- ^ Parham P, Ohta T (April 1996). "Population biology of antigen presentation by MHC class I molecules". Science 272 (5258): 67–74. Bibcode 1996Sci...272...67P. doi:10.1126/science.272.5258.67. PMID 8600539. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=8600539.
- ^ Parham P, Adams EJ, Arnett KL (February 1995). "The origins of HLA-A, B,C polymorphism". Immunol. Rev. 143: 141–80. doi:10.1111/j.1600-065X.1995.tb00674.x. PMID 7558075.
- ^ Harris EE, Hey J (March 1999). "X chromosome evidence for ancient human histories". Proc. Natl. Acad. Sci. U.S.A. 96 (6): 3320–4. Bibcode 1999PNAS...96.3320H. doi:10.1073/pnas.96.6.3320. PMC 15940. PMID 10077682. http://www.pnas.org/cgi/pmidlookup?view=long&pmid=10077682.
- ^ Harris EE, Hey J (May 2001). "Human populations show reduced DNA sequence variation at the factor IX locus". Curr. Biol. 11 (10): 774–8. doi:10.1016/S0960-9822(01)00223-8. PMID 11378388. http://linkinghub.elsevier.com/retrieve/pii/S0960-9822(01)00223-8.
- ^ Handt O, Höss M, Krings M, Pääbo S (June 1994). "Ancient DNA: methodological challenges". Experientia 50 (6): 524–9. doi:10.1007/BF01921720. PMID 8020612.
- ^ Handt O, Krings M, Ward RH, Pääbo S (August 1996). "The retrieval of ancient human DNA sequences". Am. J. Hum. Genet. 59 (2): 368–76. PMC 1914746. PMID 8755923. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1914746.
- ^ Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S (July 1997). "Neandertal DNA sequences and the origin of modern humans". Cell 90 (1): 19–30. doi:10.1016/S0092-8674(00)80310-4. PMID 9230299. http://linkinghub.elsevier.com/retrieve/pii/S0092-8674(00)80310-4.
- ^ Ingman M, Kaessmann H, Pääbo S, Gyllensten U (December 2000). "Mitochondrial genome variation and the origin of modern humans". Nature 408 (6813): 708–13. doi:10.1038/35047064. PMID 11130070.
- ^ Gonder MK, Mortensen HM, Reed FA, de Sousa A, Tishkoff SA (March 2007). "Whole-mtDNA genome sequence analysis of ancient African lineages". Mol. Biol. Evol. 24 (3): 757–68. doi:10.1093/molbev/msl209. PMID 17194802.
- ^ Kimura M (May 1954). "Process Leading to Quasi-Fixation of Genes in Natural Populations Due to Random Fluctuation of Selection Intensities". Genetics 39 (3): 280–95. PMC 1209652. PMID 17247483. http://www.genetics.org/cgi/pmidlookup?view=long&pmid=17247483.
Links
Categories:









Wikimedia Foundation. 2010.