- Chip-Sequencing
-
ChIP-Sequencing, also known as ChIP-Seq, is used to analyze protein interactions with DNA. ChIP-Seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the cistrome of DNA-associated proteins. It can be used to precisely map global binding sites for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
Contents
Uses of ChIP-Seq
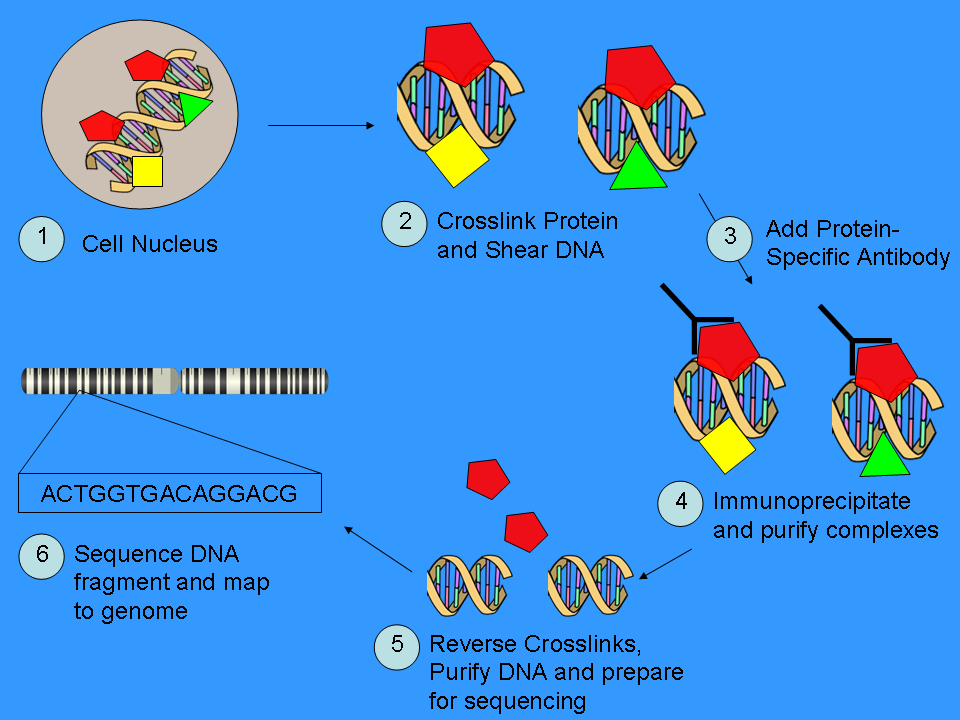
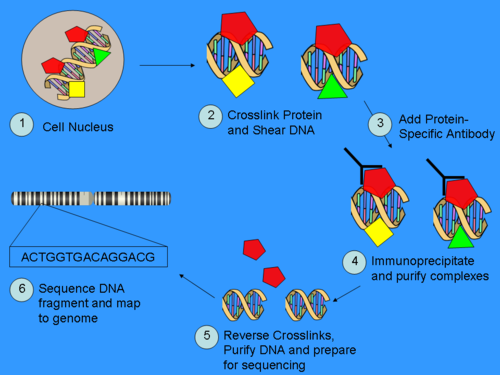 ChIP-Sequencing Workflow
ChIP-Sequencing Workflow
Chip-Seq is used primarily to determine how transcription factors and other chromatin-associated proteins influence phenotype-affecting mechanisms. Determining how proteins interact with DNA to regulate gene expression is essential for fully understanding many biological processes and disease states. This epigenetic information is complementary to genotype and expression analysis. ChIP-Seq technology is currently seen primarily as an alternative to ChIP-chip which requires a hybridization array. This necessarily introduces some bias, as an array is restricted to a fixed number of probes. Sequencing, by contrast, is thought to have less bias, although the sequencing bias of different sequencing technologies is not yet fully understood.
Specific DNA sites in direct physical interaction with transcription factors and other proteins can be isolated by chromatin immunoprecipitation. ChIP produces a library of target DNA sites bound to a target in vivo. Massively parallel sequence analyses are used in conjunction with whole-genome sequence databases to analyze the interaction pattern of any protein with DNA[1], or the pattern of any epigenetic chromatin modifications. This can be applied to the set of ChIP-able proteins and modifications, such as transcription factors, polymerases and transcriptional machinery, structural proteins, protein modifications, and DNA modifications.[2] As an alternative to the dependence on specific antibodies, different methods have been developed to find the superset of all nucleosome-depleted or nucleosome-disrupted active regulatory regions in the genome, like DNase-Seq and FAIRE-Seq.
Workflow of ChIP-Sequencing
Part 1: ChIP
ChIP is a powerful method to selectively enrich for DNA sequences bound by a particular protein in living cells. However, the widespread use of this method has been limited by the lack of a sufficiently robust method to identify all of the enriched DNA sequences. The ChIP process enriches specific crosslinked DNA-protein complexes using an antibody against a protein of interest. For a good description of the ChIP wet lab protocol see the ChIP-on-chip Wikipedia page. Oligonucleotide adapters are then added to the small stretches of DNA that were bound to the protein of interest to enable massively parallel sequencing.
Part 2: Sequencing
After size selection, all the resulting ChIP-DNA fragments are sequenced simultaneously using a genome sequencer. A single sequencing run can scan for genome-wide associations with high resolution, meaning that features can be located precisely on the chromosomes. ChIP-chip, by contrast, requires large sets of tiling arrays for lower resolution.
There are many new sequencing methods used in this sequencing step. Some technologies that analyze the sequences can use cluster amplification of adapter-ligated ChIP DNA fragments on a solid flow cell substrate to create clusters of approximately 1000 clonal copies each. The resulting high density array of template clusters on the flow cell surface is sequenced by a Genome analyzing program. Each template cluster undergoes sequencing-by-synthesis in parallel using novel fluorescently labelled reversible terminator nucleotides. Templates are sequenced base-by-base during each read. Then, the data collection and analysis software aligns sample sequences to a known genomic sequence to identify the ChIP-DNA fragments.
Sensitivity
Sensitivity of this technology depends on the depth of the sequencing run (i.e. the number of mapped sequence tags), the size of the genome and the distribution of the target factor. The sequencing depth is directly correlated with cost. If abundant binders in large genomes have to be mapped with high sensitivity, costs are high as an enormously high number of sequence tags will be required. This is in contrast to ChIP-chip in which the costs are not correlated with sensitivity.
Unlike microarray-based ChIP methods, the precision of the ChIP-Seq assay is not limited by the spacing of predetermined probes. By integrating a large number of short reads, highly precise binding site localization is obtained. Compared to ChIP-chip, ChIP-Seq data can be used to locate the binding site within few tens of base pairs of the actual protein binding site. Tag densities at the binding sites are a good indicator of protein–DNA binding affinity[3], which makes it easier to quantify and compare binding affinities of a protein to different DNA sites[4].
Current Research
- STAT1 DNA association: Recently, ChIP-Seq was used to study STAT1 targets in HeLA S3 cells. The performance of ChIP-Seq was then compared to the alternative protein–DNA interaction methods of ChIP-PCR and ChIP-chip.[5]
- Nucleosome Architecture of Promoters: Using ChIP-Seq, it was determined that Yeast genes seem to have a minimal nucleosome-free promoter region of 150bp in which RNA polymerase can initiate transcription.[6]
Conclusion
In summary, ChIP-seq offers an alternative to ChIP-chip. STAT1 experimental ChIP-seq data have a high degree of similarity to results obtained by ChIP-chip for the same type of experiment, with >64% of peaks in shared genomic regions. Because the data are sequence reads, ChIP-seq offers a rapid analysis pipeline (as long as a high-quality genome sequence is available for read mapping, and the genome doesn't have repetitive content that confuses the mapping process) as well as the potential to detect mutations in binding-site sequences, which may directly support any observed changes in protein binding and gene regulation.
Alternatives
- ChIP-on-chip
- ChIP-PET
- ChIP-PCR
Similar methods
- Sono-Seq, identical to Chip-Seq but skipping the immunoprecipitation step.
- CLIP-Seq, for finding interactions with RNA rather than DNA.
- PAR-CLIP, for identifying the binding sites of cellular RNA-binding proteins (RBPs) and microRNA-containing ribonucleoprotein complexes (miRNPs).
- RIP-Chip, same goal and first steps, but using microarray instead of sequencing
- SELEX, a method for finding a consensus binding sequence
- Competition-ChIP, to measure relative replacement dynamics on DNA.
- ChiRP-Seq to measure RNA-bound DNA and proteins.
References
- ^ Johnson DS, Mortazavi A et al. (2007) Genome-wide mapping of in vivo protein–DNA interactions. Science 316: 1497–1502
- ^ http://www.illumina.com/Documents/products/datasheets/datasheet_chip_sequence.pdf
- ^ Jothi et al. (2008) Genome-wide identification of in vivo protein–DNA binding sites from ChIP-Seq data. Nucl Acids Res 36(16) 5221–5231.
- ^ Bernstein, BE et al. (2005)Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181.
- ^ Robertson G et al.(2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods 4: 651–657.
- ^ Schmid et al. (2007) ChIP-Seq Data reveal nucleosome architecture of human promoters. Cell 131: 831–832
Categories:- Protein methods
- Molecular biology techniques
Wikimedia Foundation. 2010.