- Transthyretin-related hereditary amyloidosis
-
Transthyretin-related hereditary amyloidosis Classification and external resources OMIM 105210 eMedicine article/335301 Familial amyloid polyneuropathy (FAP), also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis or Corino de Andrade's disease,[1] is an autosomal dominant[2] neurodegenerative disease. It is a form of paramyloidosis, and was first identified and described by Portuguese neurologist Mário Corino da Costa Andrade, in the 1950s.[3] It is a fatal and incurable disease.
Contents
Characteristics
Usually manifesting itself between 20 and 40 years of age, it is characterized by pain, paresthesia, muscular weakness and autonomic dysfunction. In its terminal state, the kidneys and the heart are affected. FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motorial polyneuropathy.
Cause and genetics
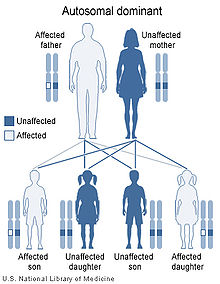 Familial amyloid polyneuropathy has an autosomal dominant pattern of inheritance.
Familial amyloid polyneuropathy has an autosomal dominant pattern of inheritance.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[4] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed]
FAP is inherited in an autosomal dominant manner.[2] This means that the defective gene responsible for the disorder is located on an autosome (chromosome 18 is an autosome), and only one copy of the defective gene is sufficient to cause the disorder, when inherited from a parent who has the disorder.
Prognosis
In the absence of a liver transplant, FAP is invariably fatal.
Epidemiology
This disease is endemic in Portuguese locations Póvoa de Varzim and Vila do Conde (Caxinas), with more than 1000 affected people, coming from about 500 families, where 70% of the people develop the illness. It was brought by Vikings from Scandinavia during the Middle Age. In northern Sweden, more specifically Piteå, Skellefteå and Umeå, 1.5% of the population has the mutated gene. There are many other populations in the world who exhibit the illness after having developed it independently.
Experimental treatments
The drug tafamidis[5] has completed a phase II/III clinical trial[6] and preliminary results suggest it slows progression of the disease.[7]
Animal models
Combined Doxycycline and TUDCA treatment (in human tolerable doses) significantly lowered fibrillar Transthyretin (TTR) amyloid deposition in transgenic TTR mice models. The authors propose this treatment in FAP, particularly in the early stages of disease.[8]
See also
References
- ^ a b Online 'Mendelian Inheritance in Man' (OMIM) 105210
- ^ a b Ando, Y.; Ueda, M. (May 2008). "Novel methods for detecting amyloidogenic proteins in transthyretin related amyloidosis". Frontiers in bioscience : a journal and virtual library 13: 5548–5558. PMID 18508604.
- ^ Andrade C (September 1952). "A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves". Brain 75 (3): 408–27. PMID 12978172. http://brain.oxfordjournals.org/cgi/pmidlookup?view=long&pmid=12978172.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) 176300
- ^ Tafamidis structure
- ^ Safety and Efficacy Study of Fx-1006A in Patients With Familial Amyloidosis.
- ^ http://protomag.ticsnetwork.com/statics/MGH_SP10_Protein_fold_F2.pdf
- ^ Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ (Jul 2010). "Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models" (Free full text). Journal of Translational Medicine 8: 74. PMC 2922089. PMID 20673327. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2922089.
External links
- Stanford University Amyloid Center
- Sousa MM, Du Yan S, Fernandes R, Guimaraes A, Stern D, Saraiva MJ (October 2001). "Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways". J. Neurosci. 21 (19): 7576–86. PMID 11567048. http://www.jneurosci.org/cgi/pmidlookup?view=long&pmid=11567048.
- Portuguese Paramyloidosis Association (in Portuguese)
- Center for study and support for Paramyloidosis (in Portuguese)
Metabolic disease: amyloidosis (E85, 277.3) Common amyloid forming proteins Systemic amyloidosis - AL amyloidosis
- AA amyloidosis
- Aβ2M/Haemodialysis-associated amyloidosis
- AGel/Finnish type amyloidosis
- AA/Familial Mediterranean fever
- ATTR/Transthyretin-related hereditary amyloidosis
Organ-limited amyloidosis HeartAANF/Isolated atrial amyloidosisCategories:- Neurological disorders
- Autosomal dominant disorders
- Skin conditions resulting from errors in metabolism
Wikimedia Foundation. 2010.