- Dihydropyrimidine dehydrogenase deficiency
-
Dihydropyrimidine dehydrogenase deficiency Classification and external resources OMIM 274270 DiseasesDB 29817 MeSH D054067 Dihydropyrimidine dehydrogenase deficiency (DPD deficiency) is an autosomal recessive[1] metabolic disorder in which there is absent or significantly decreased activity of dihydropyrimidine dehydrogenase, an enzyme involved in the metabolism of uracil and thymine.
Individuals with this condition may develop life-threatening toxicity following exposure to 5-fluorouracil (5-FU), a chemotherapy drug that is used in the treatment of cancer.[2][3] Beside 5-FU, widely prescribed oral fluoropyrimidine capecitabine (Xeloda) could put DPD-deficient patients at risk of experiencing severe or lethal toxicities as well.[4][5]
Contents
Genetics
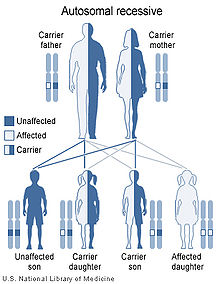 Dihydropyrimidine dehydrogenase deficiency has an autosomal recessive pattern of inheritance.
Dihydropyrimidine dehydrogenase deficiency has an autosomal recessive pattern of inheritance.
DPD deficiency is inherited in an autosomal recessive manner.[1] This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Epidemiology
Current research suggests that nearly 8% of the population has at least partial DPD deficiency. A diagnostics determination test for DPD deficiency is available and it is expected that with a potential 500,000 people in North America using 5-FU this form of testing will increase. The whole genetic events affecting the DPYD gene and possibly impacting on its function are far from being elucidated, and epigenetic regulations could probably play a major role in DPD deficiency.
It is more common among African-Americans than it is among Caucasians.[6]
References
- ^ a b Diasio RB, Beavers TL, Carpenter JT (Jan 1988). "Familial deficiency of dihydropyrimidine dehydrogenase: biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity" (Free full text). J Clin Invest. 81 (1): 47–51. doi:10.1172/JCI113308. PMC 442471. PMID 3335642. http://www.jci.org/articles/view/113308.
- ^ Van Kuilenburg AB (Mar 2006). "Screening for dihydropyrimidine dehydrogenase deficiency: to do or not to do, that's the question". Cancer investigation 24 (2): 215–217. doi:10.1080/07357900500524702. PMID 16537192.
- ^ Lee A, Ezzeldin H, Fourie J, Diasio R (Aug 2004). "Dihydropyrimidine dehydrogenase deficiency: impact of pharmacogenetics on 5-fluorouracil therapy". Clinical advances in hematology & oncology : H&O 2 (8): 527–532. ISSN 1543-0790. PMID 16163233.
- ^ Mercier C, Ciccolini J (Nov 2006). "Profiling dihydropyrimidine dehydrogenase deficiency in patients with cancer undergoing 5-fluorouracil/capecitabine therapy". Clinical colorectal cancer 6 (4): 288–296. doi:10.3816/CCC.2006.n.047. ISSN 1533-0028. PMID 17241513.
- ^ Mercier C, Ciccolini J (Dec 2007). "Severe or lethal toxicities upon capecitabine intake: is DPYD genetic polymorphism the ideal culprit?". Trends in pharmacological sciences 28 (12): 597–598. doi:10.1016/j.tips.2007.09.009. PMID 18001850.
- ^ Saif MW, Seller S, Diasio RB (Mar 2008). "Atypical toxicity associated with 5-Fluororacil in a DPD-deficient patient with pancreatic cancer. Is ethnicity a risk factor?". JOP 9 (2): 226–9. PMID 18326935. http://www.joplink.net/prev/200803/07.html.
External links
- Site dedicated exclusively to DPD Deficiency and Fluorouracil (5-FU) Toxicity related issues - dpd-deficiency.com
- Dihydropyrimidine dehydrogenase deficiency at NIH's Office of Rare Diseases
Inborn error of purine-pyrimidine metabolism (E79, 277.2) Purine metabolism AnabolismCatabolismPyrimidine metabolism AnabolismCatabolismDihydropyrimidine dehydrogenase deficiencyCategories:- Inborn errors of purine-pyrimidine metabolism
- Autosomal recessive disorders
- Genetic disorder stubs
Wikimedia Foundation. 2010.