- Tiffeneau–Demjanov rearrangement
-
The Tiffeneau-Demjanov rearrangement (TDR) is the chemical reaction of a 1-aminomethyl-cycloalkanol with nitrous acid to form an enlarged cycloketone.
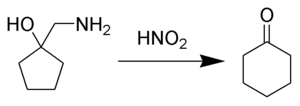
The Tiffeneau–Demjanov ring expansion, Tiffeneau–Demjanov rearrangement, or TDR, provides an easy way to increase amino-substituted cycloalkanes and cycloalkanols in size by one carbon. Ring sizes from cyclopropane through cyclooctane are able to undergo Tiffeneau–Demjanov ring expansion with some degree of success. Yields decrease as initial ring size increases, and the ideal use of TDR is for synthesis of five, six, and seven membered rings. A principal synthetic application of Tiffeneau–Demjanov ring expansion is to bicyclic or polycyclic systems. Several reviews on this reaction have been published.[1][2][3]
Contents
Discovery
The reaction now known as the Tiffeneau–Demjanov rearrangement (TDR) was discovered in two steps. The first step of occurred in 1901 when Russian chemist Nikolai Demyanov discovered that aminomethylcycloalkanes produce novel products upon treatment with nitrous acid. When this product was identified as the expanded alcohol in 1903, the Demjanov rearrangement was coined.
The Demjanov rearrangement itself has since been successfully used in industry and synthetical organic chemistry. However, its scope is limited. The Demjanov rearrangement is only best suited for expanding four, five, and six member aminomethylcycloalkanes. Moreover, alkenes and un-expanded cycloalcohols form as by-products. Yields diminish as the starting cycloalkane becomes larger.
A discovery by French scientists a few years before World War II would result in the modern TDR reaction. In 1937, Tiffeneau, Weill, and Tchoubar published in Comptes Rendus their finding that 1-aminomethylcycloahexanol converts readily to cycloheptanone upon treatment with nitrous acid.[4] Perhaps due to such a large ring being expanded, the authors did not it immediately relate it to the Demjanov rearrangement. Instead, they envisioned that their reaction was similar to one discovered by Wallack in 1906. Upon oxidation with permanganate, cycloglycols will dehydrate to yield an aldehyde via an epoxide intermediate . The authors postulated that deamination resulted in a similar epoxide intermediate that subsequently formed a ring enlarge cycloketone. However, in the time that followed, scientists began to realize that these reactions were related. By the early 1940’s, TDR was in organic vernacular. Tiffeneau’s discovery enlarged the synthetic scope of the Demjanov rearrangement as now seven and eight carbon rings could be enlarged. Since the resulting cycloketone could be easily converted to a cycloaminoalcohol again, this new method quickly became popular among organic chemists.
Basic mechanism
The basic reaction mechanism is a diazotation of the amino group by nitrous acid followed by expulsion of nitrogen and formation of a primary carbocation. A rearrangement reaction with ring expansion forms a more stable oxonium ion which is deprotonated.[5]
Early development of mechanism
Although chemists at the time knew very well what the product of a symmetrical 1-aminomethylcycloalcohol would be when exposed to nitrous acid, there was significant debate on the reaction’s mechanism that lasted up until the 1980’s. Scientists were puzzled over the array of products they would obtain when the reaction was performed on an unsymmetrical 1-aminomethylcycloalcohols and bridged cyclo-systems. Even today, experiments continue that are designed to shed light into the more subtle mechanistic features of this reaction and increase yields of desired expanded products.
In 1960, Peter Smith and Donald Baer, both of the University of Michigan, published a treatise on the TDR. Their proposed mechanism contained within provides an excellent persepective of scientist’s understanding of the TDR at that time.
The mechanism proposed by Baer and Smith was the summation of several sources of information. Since the early 1950’s, it had been postulated by many that the TDR mechanism involved a carbonium ion . However, a major breakthrough in the development of the TDR mechanism came with the improved understanding of the phenomenon behind amine groups reacting with nitrous acid. Meticulous kinetic studies throughout the late 1950’s led scientists to believe that nitrous acid reacts with an amine by first producing a nitrous acid derivative, potentially N2O3. While this derivative would prove incorrect as it relates to TDR, scientists of the time still correctly came to the conclusion that the derivative would react with the amine to produce the diazonium ion. The inferred instability of this diazonium ion gave solid evidence for the existence of a carbocation in the TDR mechanism.
Another piece of information that had implications in the mechanism of the TDR was the simple fact that the reaction proceeds more easily with the conditions discovered by Tiffeneau. By placing an alcohol on the carbon on the reagent, reaction rates and yields are much improved to those of the simple Demjanov rearrangement. Moreover, few unwanted by products are formed, such as olefins. These aforementioned observations were the center around which Smith and Baer’s mechanism was constructed. It is easy to see that hydrogen’s presence would mean that hydride shifts would occur in competition with carbon shifts during the course of the reaction. Moreover, this shift is likely as it would move place positive charge from a 1° carbon to a 3° carbon. In a mildly basic solvent such as water, this new intermediate could easily produce an olefin via an E1-like reaction.
Such olefins are typically seen in simple Demjanov rearrangements but are not seen in the TDR. The alcohol’s presence explains how this E1 reaction does not occur. Moreover, having an alcohol present puts the developing positive charge of the ring enlarged intermediate next to an oxygen. This would be more favorable than hydrogen as oxygen can lend electron density to the carbonium ion via resonance.
This again favors ring expansion and is another caveat that shows how it incorporates higher yields of the TDR over the Demjanov rearrangement.
Smith and Baer’ mechanism was also able to account for other observations of the time. Tiffeneau-Demjanov rearrangements of1-aminomethylcycloalkanols with alkyl substitutions on the side aminomethyl chain had been accomplished by many scientists before 1960. Smith and Baer investigated how such substitution affects the TDR by synthesizing various 1-hydroxycyclohexylbenzyl-amines and exposing them to TDR conditions .
Seeing as six member rings are routinely enlarged by the TDR, one might expect the reaction to occur. However instead of the anticipated ring enlargements, only diols are seen as products. Interestingly enough, five member analogues to the above substituted reagents enlarge under TDR conditions. Alkyl substitutions as opposed to aryl substitutions result in diminished TDRs. Smith and Baer assert that these observations support their mechanism. Since substitution stabilizes the carbonium ion after daminification, the resulting carbonium ion is more likely to react with a nucleophile present (water in this case) and not undergo rearrangement. Five member rings rearrange due to the ring strain encouraging the maneuver. This strain makes the carbocation unstable enough to cause a carbon to shift.
Problems with the early mechanism
As definitive as Smith and Baer’s early mechanism seems, there are several phenomena that it did not account for. The problem with their mechanism mainly focused around TDR precursors that have alkyl substituents on the ring. When said substituent is placed on the ring as to make the molecule still symmetric, one product is formed upon exposure to TDR conditions. However, if the alkyl is placed on the ring as to make the molecule unsymmetric, several products could form.
The principal method for synthesizing the starting amino alcohols is through the addition of cyanide anion to a cyclic ketone. The resulting hydroxynitrile is then reduced, forming the desired amino alcohol. This method forms diastereomers, possibly affecting the regioselectivity of the reaction. For nearly all asymmetric precursors, one product isomer is formed preferentially to another. As TDR was routinely being used to synthesize various steroids and bicyclic compounds, their precursors were rarely symmetric. As a result, a lot of time was spent identifying and separating products. At the time, this phenomenon baffled chemists. Due to spectroscopic and separation limitations, it was very difficult for scientists to probe this caveat of the TDR in a sophisticated way. However, most believed that what was governing preferential product formation involved the migratory aptitudes of competing carbons and/or steric control. Migratory aptitude made reference to the possibility that the preferred product of the reaction was the result of an initial stability of one carbon migrating in preference to another. This possibility was more the belief and subject of research of earlier scientists, including Marc Tiffeneau himself. However in the early 1960s, more and more scientists were starting to think that steric factors were the driving force behind the selectivity for this reaction.
Sterics and Stereochemistry in the mechanism
As chemists continued to probe this reaction with more and more advanced technology and methods, other factors began to be tabled as possibilities for what was controlling product formation of unsymmetrical amino alcohols. In 1963, Jones and Price of the University of Toronto demonstrated how remote substituents in steroids play a role in product distribution . In 1968, Carlson and Behn of the University of Kansas discovered that experimental conditions also play a role. These latter scientists established that in ring extension via the TDR, initial temperature and concentration of reagents all played a role in eventual product distribution . Indeed, other avenues of the TDR were being explored and charted.
However, Carlson and Behn did manage to report a significant breakthrough in the realm of sterics and migratory aptitudes as they relate to the TDR. As it might be expected based on electronic reasoning, the more highly substituted carbon should migrate preferentially to a less substituted carbon. However, this is not always seen and often accounts of migratory aptitudes show fickle preferences. Thus, the authors assert that such aptitudes are of little importance. Stericlly, thanks chiefly to improved spectroscopic methods, they were able to confirm that having the amine group equatorial to the alkane ring corresponded to drastically different product yields.
According to the authors, the preferential formation of D from A does not reflect a preferred conformation of A. Their modeling indicates that both A and B are initially just as likely to become C. He concludes that there must be a steric interaction to develop in the transition state during migration that makes A preferentially form D when exposed to the TDR conditions. The idea that sterics played a factor during migration and was not a factor just at the beginning to the reaction, was new. Carlson and Behn speculate that the factor might lay in transannular hydrogen interactions along the path of migration. Their modeling suggested that this interaction may be more severe for A forming C. However, they are not certain enough to offer this as a definitive explanation as they concede that more subtle conformational and/or electronic effects could be at work as well.
At this point, the mechanism proposed by Smith and Baer seemed to be on its way out. If steric interactions relating to carbon migration during the reaction’s transition state were important, this did not support the carbocation envisioned by Smith and Baer. Research around bi-cyclics during the 1970’s would shed even more light into the TDR mechanism. In 1973, McKinney and Patel of Marquette University published an article in which they used the TDR for expanding Norcamphor and Dehydronorcamphor . Two of their observations are important. One centers on the expansion of exo and endo-2-norbornylcarbinyl systems.
One might expect in (I) that A would migrate in preference to B seeing as such a migration would place the developing charge on a 2° carbon and pass the specie through a more favorable chair-like intermediate. This is not seen. Only 38% of the product exhibits A migration. To account for why A migration is not dominant in the expansion of I, the authors assert a least movement argument . Simply put, the migration of the non-bridgehead carbon provides for the least amount of total atom movement, something that plays into the energetics of the reaction. This least movement consideration would prove important in the TDR mechanism as it accounts for products with intermediates passing through unfavorable conformations.
However, McKinney and Patel also confirm that traditional factors such as developing positive charge stability still play a crucial role in the direction of expansion. They accomplish this by expanding 2-norbornenyl carbinyl systems.
By adding a simple double bond to these systems, the authors see a significant increase in the migration of the bridgehead carbon A (50% in this case.) The authors attribute this jump in migration to the fact that this bride carbon migrating allows the developing positive charge to be stabilizing by resonance contributed by the double bond .Therefore, carbocation/ positive charge effects can not be ignored in the discussion of the factors influencing product distribution.
Later mechanistic studies
As evidence continued to mount during the years after Smith and Baer’s publication in 1960, it was obvious that the TDR mechanism would need revisiting. This new mechanism would have to de-stress the carbocation as there are other factors that influence ring expansion. Orientation of the developing diazonium ion, the possibility of steric interactions during the reaction, and atomic movement would all have to be included. In 1982, Cooper and Jenner published such a mechanism .[citation needed] Their mechanism has stood to this day as the current understanding of the TDR .
The most obvious departure from Smith and Baer’s mechanism is that Cooper and Jenner represent the diazonium departure and subsequent alkyl shift as a concerted step. Such a feature allows for sterics, orientations, and atomic movement to be factors. However, distribution of positive charge is still important in this mechanism as it does explain much of the observed behavior of the TDR. Another observation that should be made is that there is no preference given to these aforementioned factors in the mechanism. That is to say, even today it is very difficult to predict which carbon will migrate preferentially. Indeed, the TDR has become more useful as spectroscopic and separation techniques have advanced. Such advancements allows for the quick identification and isolation of desired products.
Since the mid 1980’s, most organic chemists have resigned themselves to accepting the fact that the TDR is governed by several factors that often seem fickle in importance. As a result, much research is now being directed towards the development of techniques to increase migration of a specific carbon. One example of such an effort has recently come out of the University of Melbourne.
Noting that group 4 metal substituents can stabilize positive charge that is β to them, Chow, McClure, and White attempted to use this to direct TDRs in 2004.[citation needed] They hypothesized that placing a silicon trimethyl group β to a carbon that can migrate would increase such migration .
Their results show that this does occur to a small extent. The authors believe that the reason why the carbon migration increases only slightly is that positive charge is not a large factor in displacing the diazonium ion. Since this ion is such a good leaving group, it requires very little ‘push’ from the developing carbon-carbon bond. Their results again highlight the fact that multiple factors determine the direction of carbon migration.
References
- ^ Smith, P. A. S.; Baer, D. R. Org. React 1960, 11, 157–188. (Review)
- ^ Coveney, D. J. Comp. Org. Syn. 1991, 3, 781–782. (Review)
- ^ Fattori, D. et al. Tetrahedron 1993, 49, 1649. (Review)
- ^ Marc Tiffeneau; Paul Weill; Bianca Tchoubar (1937). "Isomérisation de l'oxyde de méthylène cyclohexane en hexahydrobenzaldéhyde et désamination de l'aminoalcool correspondant en cycloheptanone". Comptes Rendus 205: 54–56. http://gallica.bnf.fr/ark:/12148/bpt6k3157c.image.f54.
- ^ Jack Li, Jie. (2006). Name Reactions (Third ed.). Berlin: Springer.
See also
Categories:- Rearrangement reactions
- Name reactions

Wikimedia Foundation. 2010.