- Divinylcyclopropane-cycloheptadiene rearrangement
-
The divinylcyclopropane-cycloheptadiene rearrangement is an organic chemical transformation that involves the isomerization of a 1,2-divinylcyclopropane into a cycloheptadiene or -triene. It is conceptually related to the Cope rearrangement, but has the advantage of a strong thermodynamic driving force due to the release of ring strain.[1]
Contents
Introduction
In 1960, Vogel discovered that 1,2-divinylcyclopropane rearranges to cycloheptan-1,4-diene.,[2] After his discovery, a series of intense mechanistic investigations of the reaction followed in the 1960s, as researchers realized it bore resemblance (both structural and mechanistic) to the related rearrangement of vinylcyclopropane to cyclopentene. By the 1970s, the rearrangement had achieved synthetic utility[3] and to this day it continues to be a useful method for the formation of seven-membered rings. Variations incorporating heteroatoms have been reported (see below).
(1)
Advantages: Being a rearrangement, the process exhibits ideal atom economy. It often proceeds spontaneously without the need for a catalyst. Competitive pathways are minimal for the all-carbon rearrangement.
Disadvantages: The configuration of the starting materials needs be controlled in many cases—trans-divinylcyclopropanes often require heating to facilitate isomerization before rearrangement will occur. Rearrangements involving heteroatoms can exhibit reduced yields due to the formation of side products.
Mechanism and Stereochemistry
Prevailing Mechanism
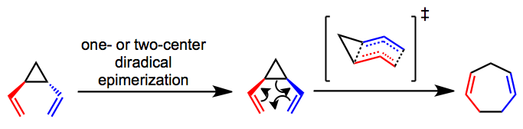
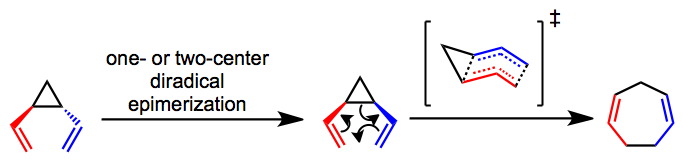
The primary debate concerning the mechanism of the rearrangement centers on whether it is a concerted (sigmatropic) or stepwise (diradical) process. Mechanistic experiments have shown that trans-divinylcyclopropanes epimerize to the corresponding cis isomers and undergo the rearrangement via what is most likely a concerted pathway.[4][5] A boat-like transition state has been proposed and helps explain the observed stereospecificity of the process. Whether the initial epimerization of trans substrates occurs via a one- or two-center process is unclear in most cases.
(2)
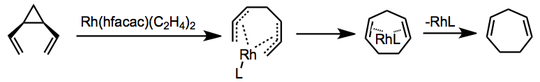
Transition-metal-catalyzed versions of the rearrangement are known, and mechanisms vary. In one example employing rhodium bis(ethylene) hexafluoroacetylacetonate, coordination and formation of a bis-π-allyl complex precede electrocyclic ring closure and catalyst release.[6]
(3)
Stereoselective Variants
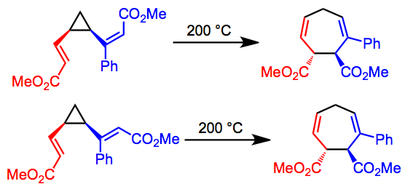
Reactions of divinylcyclopropanes containing substituted double bonds are stereospecific with respect to the configurations at the double bonds—cis,cis isomers give cis products, while cis,trans isomers give trans products. Thus, chiral, non-racemic starting materials give rise to chiral products without loss of enantiomeric purity. In the example below, only the isomers depicted were observed in each case.[7]
(4)
Scope and Limitations
A wide variety of divinylcyclopropanes undergo the title reaction. These precursors have been generated by a variety of methods, including the addition of cyclopropyl nucleophiles (salts of lithium,[8] or copper[9]) to activated double or triple bonds, elimination of bis(2-haloethyl)cyclopropanes[10] and cyclopropanation.[11]
In the example below, cuprate addition-elimination generates the transient enone 1, which rearranges to spirocycle 2.
(5)

Organolithiums can be employed in a similar role, but add in a direct fashion to carbonyls. Products with fused topology result.[12]
(6)

Rearrangement after elimination of ditosylates has been observed; the chlorinated cycloheptadiene thus produced isomerizes to conjugated heptadiene 3 during the reaction.[10]
(7)

Cyclopropanation with conjugated diazo compounds produces divinylcyclopropanes that then undergo rearrangement. When cyclic starting materials are used, bridged products result.[13]
(8)

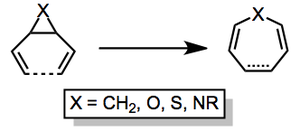
Substrates containing three-membered heterocyclic rings can also undergo the reaction. cis-Divinylepoxides give oxepines at elevated temperatures (100 °C). trans Isomers undergo an interesting competitive rearrangement to dihydrofurans through the intermediacy of a carbonyl ylide[14] and the same ylide intermediate has been proposed as the direct precursor to the oxepine product 4.[15] Conjugated dienyl epoxides form similar products, lending support to the existence of an ylide intermediate.[16]
(9)

Divinyl aziridines undergo a similar suite of reactions providing azepines or vinyl pyrrolines depending on the relative configuration of the aziridine starting material.[17] Divinyl thiiranes can provide thiepines or dihydrothiophenes, although these reactions are slower than those of the corresponding nitrogen- and oxygen-containing compounds.
Synthetic Applications
The earliest observation of a cycloheptadiene via the title rearrangement was made by Baeyer in his synthesis of eucarvone from carvone hydrobromide.[18] Mechanistic studies revealed that the rearrangement did indeed proceed via a concerted, Cope-type mechanism.[19]
(10)

In the Eschenmoser synthesis of colchicine, the rearrangement is used to form the seven-membered ring of the target.[20]
(11)

A racemic synthesis of sirenin employs a Wittig reaction to form the key divinylcyclopropane. Hydrogenation of the rearrangement product afforded the target.[21]
(12)

Experimental Conditions and Procedure
Typical Conditions
Typically, the rearrangement is carried out just after the formation of the divinylcyclopropane, in the same pot. Heating is sometimes necessary, particularly for trans substrates, which must undergo epimerization prior to rearrangement. With enough energy to surmount activation barriers, however, the isomerization is usually very efficient.
Example Procedure[22]
(13)

To a cold (–78°) stirred solution of lithium diisopropylamide (1.4–1.5 mmol/mmol of ketone) in dry THF (4 mL/mmol of base) under an atmosphere of argon was added slowly a solution of n-butyl-trans-2-vinylcyclopropyl ketone (1.19 mmol) in dry THF (1 mL/mmol of ketone), and the resulting solution was stirred at –78° for 45 minutes. A solution of freshly sublimed tert-butyldimethylsilyl chloride (1.6 mmol/mmol of ketone) in dry THF (1 mL/mmol of chloride) was added, followed by dry HMPA (0.5 mL/mmol of ketone). The solution was stirred at –78° for 15 minutes and at room temperature for 2–3 hours, and then it was partitioned between saturated aqueous sodium bicarbonate and pentane (10 mL and 20 mL/mmol of ketone, respectively). The aqueous phase was washed twice with pentane. The combined extract was washed four times with saturated aqueous sodium bicarbonate and twice with brine, and then dried (MgSO4). Removal of the solvent, followed by bulb-to-bulb distillation of the remaining oil, gave the corresponding silyl enol ether as a colorless oil that exhibited no IR carbonyl stretching absorption. Thermolysis of the silyl enol ether was accomplished by heating (neat, argon atmosphere) at 230° (air-bath temperature) for 30–60 minutes. Direct distillation (140–150°/12 torr) of the resultant materials provided the cycloheptadiene in 85% yield: IR (film) 1660, 1260, 840 cm–1; 1H NMR (CDCl3) δ 0.09 (s, 6H), 0.88 (s, 9H), 0.7–2.75 (m, 14H), 4.8 (t, 1H, J = 5.5 Hz), 5.5–5.9 (m, 2H).
References
- ^ Hudlicky, T.; Fan, R.; Reed, J. W.; Gadamasetti, K. G. Org. React. 1992, 41, 1-133. doi: (10.1002/0471264180.or041.01)
- ^ Vogel, E. Angew. Chem. 1960, 72, 4.
- ^ Wender, P. A.; Eissenstat, M. A.; Filosa, M. P. J. Am. Chem. Soc. 1979, 101, 2196.
- ^ Arai, M.; Crawford, R. J. Can. J. Chem. 1972, 50, 2158.
- ^ Baldwin, J. E.; Fleming, R. H.J. Am. Chem. Soc. 1973, 95, 5256.
- ^ Alcock, N. W.; Brown, J. M.; Conneely, J. A.; Stofko, Jr., J. J. J. Chem. Soc., Chem. Commun., 1975, 234.
- ^ Brule, D.; Chalchat, J. C.; Vessiere, R. Bull. Soc. Chim. Fr. 1978, No. 7-8, II-385.
- ^ Wender, P. A.; Filosa, M. P. J. Org. Chem. 1976, 41, 3490.
- ^ Marino, J. P.; Browne, L. J. Tetrahedron Lett. 1976, 3245.
- ^ a b Muller, P.; Rey, M. Helv. Chim. Acta, 1982, 65, 1191.
- ^ Hudlicky, T.; Rulin, F.; Lovelace, T.; Reed, J. W. in Studies in Natural Product Chemistry, Atta-ur-Rahman, Ed., Elsevier, Amsterdam, 1989, Part B, p. 3.
- ^ Wender, P. A.; Filosa, M. P. J. Org. Chem. 1976, 41, 3490.
- ^ Davies, H. M. L.; Clark, D. M.; Smith, T. K. Tetrahedron Lett. 1985, 26, 5659.
- ^ Pommelet, J. C.; Manisse, N.; Chuche, J. Tetrahedron, 1972, 28, 3929.
- ^ Braun, R. A. J. Org. Chem. 1963, 28, 1383.
- ^ Eberbach, W.; Roser, J. Tetrahedron Lett. 1987, 28, 2685.
- ^ Manisse, N.; Chuche, J. Tetrahedron, 1977, 33, 2399.
- ^ Baeyer, A. Ber. 1894, 27, 810; ibid. 1898, 31, 2067.
- ^ Vogel, E.; Ott, K.-H.; Gajek, K. Justus Liebigs Ann. Chem. 1961, 644, 172.
- ^ Schreiber, von J.; Leimgruber, W.; Pesaro, M.; Schudel, P.; Threlfall, T.; Eschenmoser, A. Helv. Chim. Acta 1961, 44, 540.
- ^ Jaenicke, L.; Akintobi, T.; Muller, D. G. Angew. Chem., Int. Ed. Engl. 1971, 10, 492.
- ^ Piers, E.; Burmeister, M. S.; Reissig, H. U. Can. J. Chem. 1986, 64, 180.
Categories:- Organic reactions
- Cyclopropanes



Wikimedia Foundation. 2010.